Review Article - (2023) Volume 7, Issue 2
Therapeutic Capability of Selected Medicinal Plants Bioactive Constituents against the Mutant Ovarian TP53 Gene; A Computational Approach
Koyad Raheem*
Department of Oncology, Adekunle Ajasin University, Ondo, Akungba, Nigeria
*Correspondence:
Koyad Raheem, Department of Oncology, Adekunle Ajasin University, Ondo, Akungba,
Nigeria,
Email:
Received: 31-May-2023, Manuscript No. IPRJO-22-14094;
Editor assigned: 02-Jun-2023, Pre QC No. IPRJO-22-14094 (PQ);
Reviewed: 16-Jun-2023, QC No. IPRJO-22-14094;
Revised: 21-Jun-2023, Manuscript No. IPRJO-22-14094 (R);
Published:
28-Jun-2023, DOI: 10.36648/iprjo.23.7.12
Abstract
Background: The pivotal role of mutant P53 protein in ovarian cancer and the efficacy of natural compounds in cancer treatment necessitated the current study to identify novel mutant P53 modulators from medicinal plants. Homology modelling was deployed to assemble the 3D structure of the mutant P53 protein from its amino acid sequences, while Findsitecom2.0 was used to predict the active binding site of the mutant P53 protein model. The bioactive constituents obtained from seven plants were used as ligands and docked against the binding pocket of mutant P53 protein. Autodock tools, PyRx and discovery studio, were used to prepare the protein, dock the ligands and visualize the complexes, respectively. Thiotepa and germcitabine were used as reference drugs. The hit compounds were selected based on their highest binding affinity and further analyzed to identify their pharmacokinetic properties and acute rat toxicity using SWISSADME and Gusar, with their electronic properties calculated using the Density Functional Theory (DFT) method.
Results: Screening results of 50 bioactive phytochemicals confirmed that 15 leads showed superior binding energies to mutant P53 as compared to the standard FDA approved drugs (thiotepa and germcitabine with binding scores of -3.5 and -5.4, respectively). After considering their drug like, pharmacokinetic properties and acute toxicity prediction, four major hits (Morusin, Irinotecan, Rubitecan and 10-hydroxycamptothecin) were identified to have minimal toxicities and are safe to be used. The DFT calculations showed regions of the molecules prone to electrophilic and nucleophilic attacks.
Conclusions: The current study revealed drug like compounds that can serve as potential modulators of mutant P53 in ovarian cancer treatment.
Keywords
Ovarian cancer; Mutant P53; Homology modelling; Molecular docking; Medicinal plants;
Density Functional Theory (DFT)
Abbreviations
OC : Ovarian Cancer; HGSOC: High-Grade Serous Ovarian Carcinoma; LGSOC: Low-grade Serous
Ovarian Carcinoma; EnOC: Endometrioid Ovarian Cancer; OCCC: Ovarian Clear Cell Carcinoma; VEGF:
Vascular Endothelial Growth Factor; MPS: Massive Parallel Sequencing; BLAST: Basic Local Alignment Search
Tool; HMMs: Hidden Markov models; VADAR: Volume, Area, Dihedral Angle Reporter; RESPROX: Resolution by
proxy; RMSD: Root Mean Square Deviation; ADME: Absorption Distribution Metabolism Excretion; SMILES:
Simplified Molecular-Input Line-Entry System; GUSAR: General Unrestricted Structure-Activity Relationships; LD50:
Median Lethal Dose; QSAR: Quantitative Structure-Activity Relationship; FMOs: Frontier Molecular Orbitals; MEP:
Molecular Electrostatic Potential; B3LYP: Becke, 3-parameter, Lee-Yang-Parr; HOMO: highest occupied
molecular orbital; LUMO: lowest unoccupied molecular orbital; GMQE: Global Model Quality Estimation; EA:
Electron Affinity; MEP: Molecular Electrostatic Potential.
INTRODUCTION
Ovarian Cancer (O.C.) is widely recognized as the most lethal gynecologic malignancy, with an estimated 295,414 newly diagnosed cases and 184,799 deaths worldwide in 2018. Due to the female anatomical features, there is a great challenge with respect to early diagnosis; hence, most women with ovarian cancer are diagnosed at the metastatic stage. Epithelial Ovarian cancer can be classified into five major subtypes: High Grade Serous Ovarian Carcinoma (HGSOC), Low Grade Serous Ovarian Carcinoma (LGSOC), Endometrioid Ovarian Cancer (EnOC), Ovarian Clear Cell Carcinoma (OCCC) and Mucinous [1]. The HGSOC are the predominant form, accounting for almost 70% of cases. Several risk factors associated with ovarian cancer include pregnancy, breastfeeding, tubal ligation, female sterilization and oral contraceptives. Other notable predisposition factors are genetics (inheritable BRCA 1/2 gene mutations) and lifestyle (smoking and unhealthy diets).
P53 is a crucial gene that produces a protein responsible for the regulation of cell division, proliferation and differentiation [2]. P53 gene resides within chromosome 17, consisting of 11 exons and 13 introns and 7 domains, including the N-terminal transactivation domain, crucial for activating pro-apoptotic genes. It regulates cellular activities such as cell division and growth, promotes apoptosis and cell cycle blockade and inhibits VEGF-induced vascularization (Vascular Endothelial Growth Factor), metastasis and angiogenesis. Other roles associated with p53 include DNA damage correction, autophagy, proliferation, inhibition of epithelial to mesenchymal transition and other subliminal activities that precede metastasis. Mutations to the codon 47 of the N-terminal have been implicated in the decreased apoptotic ability of this gene. Adjacent to the C-terminal of the wild-type p53 gene is the DNA binding domain a highly conserved sequence, which has been responsible for about 80% of all p53 mutations [3]. Gene knock in studies in mouse models further established that mutations in these hotspots resulted in a loss of function and a decreased tumour suppressive activity of the wild type p53 gene [4].
p53 is the most mutated gene responsible for over 50% of malignant outgrowths in humans. Genome association studies of ovarian cancer cells via Massive Parallel Sequencing (MPS) have implicated p53 mutation to be the causal factor of over 95% of severe ovarian cancer, with immuno histochemical assay being able to detect a high level of expression of mutated p53 having missense mutations. Early independent studies confirmed that this mutation occurs within exons 2-11 of the p53 gene, thereby leading to the loss of its onco suppressive potential.
Dysregulated activities by mutant p53 protein in cancer cells lead to the activation of survival pathways in cancer, such as the PI3K/mTOR signalling, hence making the pathway a potential target for molecular therapies [5]. Another possible therapeutic strategy involves limiting interactions between wtp53 protein and MDM2/MDM4 during DNA damage to facilitate repair of damaged genes, induce apoptosis and inhibit tumour growth. Natural products obtained from plants have been explored in the treatment of ovarian cancer. Morus alba, a typical medicinal plant found in China, contains bioactive compounds such as Kuwanon G, Moracin M, Mulberrocide, etc., which have some medicinal value. Studies have reported the anti-inflammatory anti-bacterial anti-diabetic and anticancer activities of M. alba. One of the anti-cancer mechanisms of M. alba involves the inhibition of focal adhesion kinase and Src activity in macrophages associated with tumors.
Camptotheca acuminata is a member of the Nyssaceae family of plants from China, mainly used for ornamental purposes [6]. Camptothecin, the most consequential product of C. acuminata, has been reported to show strong anti-cancer activity via inhibition of topoisomerase 1 by preventing the re-joining step of the cleavage and re-ligation of the double stranded DNA beyond its potent anti-cancer activity, there are emerging reports suggesting the antibacterial and anti-viral activity of C. acuminata. Established further that Camptothecin impacted negatively on viral replication of FAdv4 infected Leghorn male hepatocellular cells in a dose dependent manner, thereby decreasing the viral copy number and viral Hexon protein expression. Aspalathus linearis is a woody shrub with bright green and needle shaped leaves with an average height of 1.2 m-2 m.
Aspalathus linearis contains two unique flavonoid compounds: Aspalathin and aspalalinin. Other flavonoids have been characterized from Aspalathus linearis, including chrysoeriol, luteolin and luteolin-7-o-glucoside, quercetin, quercetin-3- orobinoside, hyperoside, isoquercitrin and rutin. Quercetin has been recognized for its anti-tumour, anti-proliferative and proapoptotic effects [7]. A study shows that Quercetin slowed down and possibly reversed metastasis by interfering with uPA/uPAR systems, AMPKα, NF-κβ, ERK1/2 and PKC-δ regulation.
This study evaluated the inhibitory properties of selected phytochemicals from various plants using an in-silico approach to target a mutant model of the p53 protein in ovarian cancer.
Materials and Methods
Template Identification, Binding Site Prediction and Protein Homology Modelling
The mutant P53 gene with the sequence ID: rs28934578 was retrieved from the NCBI web serve and the FASTA file of the sequence was downloaded. The complete mutant P53 protein sequence, which consists of 393 amino acids and has a calculated molecular weight of 43.653kDa, was retrieved from the UniprotKB database accession number P04637. The homology modelling was performed based on the following step; template search, sequence alignment, model building and evaluation. Template search for the target sequence was carried out with Basic Local Alignment Search Tool (BLAST) and Hidden Markov Models (HMMs): ‘HMM-HMM based lightning fast iterative sequence search’ against the updated SWISSMODEL library [8]. Using the template with the highest sequence identity and highest coverage, the protein structure of the query sequence was modelled. Accordingly, the crystal structure of cellular tumor antigen p53 (PDB ID: 3Q05.1), which has 92.07% sequence identity to mutant p53, was selected as the template (based on its high resolution) to predict the threedimensional structure of mutant P53. The binding site was predicted by FindsiteCombo2.0, maintained by the Skolnick research group at the Georgia institute of technology.
3D structure of mutant p53 was modelled using the SWISSMODEL tool in the ExPASy bioinformatics resource portal and viewed using Swiss PDB Viewer v 4.0.1 software.
Quality Assessment of the Model
The accuracy of the 3D model of mutant P53 was assessed by three online tools: PROCHECK, VERIFY 3D and ERRAT, available from the Structural Analysis and Verification Server (SAVES). PROCHECK was used to assess the stereochemical quality of the protein structure [9]. ERRAT computes and statistically compares the interaction of non-bound heavy atom pairs with standards. It is accessible at molecular biology institute, University of California, Los Angeles. VERIFY 3D program analyzed the compatibility of an atomic model (3D) with its amino acid sequence (1D) to assess the 3D protein structure. The model protein was validated using ProTSAV, a meta server performing consensus quality assessment using different validation tools including Verify3D, MolProbity, Procheck, ProSA, ERRAT, ProQ, dDFire, Naccess and D2N [10]. The overall quality of the modelled is given as a ProTSAV score which is a cumulative estimate of the individual protein quality score from the different validation tools. Lastly, quantitative analysis of the final model was performed using Volume, Area, Dihedral Angle Reporter (VADAR).
Receptor Preparation, Ligand Selection, Molecular Docking
Receptor preparation: The crystal structure of the p53 cellular tumor antigen (PDB ID: 3Q05) was extracted from the RCSB protein data bank. The Zn2+ ligand was eliminated from the protein. The chain A residues were isolated from other chains (B, C, D, K and L) residues in the PyMOL environment [11]. The protein structure was modified by adding Kollman charges and polar hydrogen atoms using the BIOVIA discovery studio and autodock tools. Water molecules were also eliminated during this process. The protein was further converted to autodock vina format (PDBQT) in the PyRx environment.
Ligand selection and preparation: In previous literature, we looked at seven plants that have been shown to be effective against ovarian cancer and their phytochemicals were retrieved from PubChem databases [12]. A total of 50 phytochemicals were retrieved from these plants. These ligands’ MOL SDF format was converted to a PDBQT file using PyRx to produce atomic coordinates and energy was reduced using PyRx’s optimization technique with the force field set to MMFF94.
Molecular docking: The ligands and protein were docked using Autodock Vina, which is included in the PyRx package [13]. PyRx is a computer based drug discovery program that may be used to screen libraries of compounds against prospective therapeutic targets. The docking was carried out utilizing Vina Wizard once both the ligands and the target had been prepared [14]. The 3Q05 protein structure was used as an input receptor while the plants’ metabolites as ligands. With the following parameters, a grid box was set up to cover the active parts of the protein structure: center (X, Y, Z)=(-60.17, -6.10, -12.14), dimensions in Angstrom (A’) (X, Y, Z)=(31.74, 32.21, 25.0) with an exhaustiveness of 8 and then vina was used to finish the docking procedure. The docked complex’s binding energy and Root Mean Square Deviation (RMSD) were obtained [15]. Csv format the top ranked metabolites were then selected based on their high binding energies ranging from -7.0 and below. The discovery studio software was then used to visualize the docked complex. Thiotepa and Gemcitabine were used as standards for the docking.
Pharmacokinetic Properties of the Compounds
The drug library was subjected to Absorption Distribution Metabolism Excretion (ADME) study using the Swiss ADME program to find the best soluble, druggable, lead like and non-violating drug compounds [16]. For each ligand, an input file containing their SMILES (Simplified Molecular Input Line Entry System) was obtained.
Acute toxicity prediction: The GUSAR (General Unrestricted Structure Activity Relationships) online module was used for the in silico acute toxicity prediction. The GUSAR software predicts toxicity based on a database of approximately 10,000 chemical structures in the software library [17]. The median lethal dose (LD50) (log10 (mg/kg) of the chemical entity was predicted using QSAR (Quantitative Structure-Activity Relationship) analysis. The analysis considers the various oral, subcutaneous, intravenous and intraperitoneal routes of administration.
LD50 predictions using GUSAR: The acute toxicity of the lead compounds (ligands) after ADME screening was predicted using the free online GUSAR software. The LD50 values for rats with different routes of administration, such as Intravenous (IV), oral, Intraperitoneal (IP) and Subcutaneous (SC) [18]. The predicted values showed that most of the lead compounds were Class V compounds (>2000 mg/kg) based on the GHS 5 category hazard classification.
Density Functional Theory (DFT) Calculation
The stability and reactivity of the lead compounds were estimated in Schrodinger materials science (version 3.9), which accommodates the Jaguar fast engine [19]. The Frontier Molecular Orbitals (FMOs) and Molecular Electrostatic Potential (MEP) were estimated using 6-31G** as the basis set and Becke’s three parameter exchange potential and Lee Yang Parr correlation (B3LYP) as the level of functional theory [20]. Intrinsic reactivity descriptors such as frontier molecular orbitals Highest Occupied Molecular Orbital (HOMO) and Lowest Unoccupied Molecular Orbital (LUMO) energy, chemical hardness, chemical softness, ionization potential, electronegativity and electron affinity were calculated [21]. The following equation can be used to define the global descriptors.

Results
Homology Modelling and Validation
The crystallized 3D structure of the mutant P53 protein was
modelled due to its absence on the protein data bank.
Homology modelling is a predictive method which is based on
similar templates and it was used to model the structure of the
protein. Previously reported that the validity and accuracy of a
predicted model largely depend on the level of similarity within
the template structures, as an intense template search was
carried out prior to protein modelling. HHblits found a total of
459 templates and the BLAST search algorithm generated 49
templates that match the target sequence. The cellular tumor
antigen P53 with PDB ID: 3QO5 showed the highest amino acid
sequence similarity with a sequence identity of 92.70%, 2.40 Ǻ;
as such was deemed the best fitting template for modelling the
3D structure of mutant P53 (Figure 1). The model protein has
a Global Model Quality Estimation (GMQE) value of 0.51 and
a QMEAN score of 0.74, indicating that the modelled
structure is reliable and of very good quality. Therefore, the
three-dimensional structure of the model protein built by
homology modelling conforms to the quality model reliability
and realistic requirement (Figure 2). The model consists of 234 amino acid residues [22]. Further analysis was carried out to
ascertain its good quality. In the Ramachandran plot, 88.6% of
the residues are within the favoured region and 11.4% lie
within the allowed region, while there are no residues (0.0%) in
the disallowed regions (Figure 3). It is proposed that a good
reliable model should have more than 80% of its residues in
the favored region. Subsequently, the final model developed
was validated using different external web servers. In ProSA
the Z-score is within the normal range of experimentally solved
structures. In ERRAT, it is known as the “overall quality factor
“for non-bonded atomic interactions, with higher scores
indicating higher quality [23]. The generally accepted range is
>50 for a high-quality model. The overall quality of the 3D
model is 93.3% (Figure 4). While ERRAT server is arguably one
of the best protein quality check servers, like other protein
quality predictors, it is not individually comprehensive; hence, a
protein quality meta server which combines diverse quality
assessment programmes (including Verify3D, MolProbity,
Procheck, ProSA, ERRAT, ProQ, dDFire, Naccess and D2N) and
outperforms their individual server accuracies were used to
give a more robust protein quality estimate [24]. Interestingly,
most of the protein structure checks tools gave an RMSD value
in the range of 1-3 A, which suggests a good overall quality of
the modelled structure (Figures 5 and 6).

Figure 1: The three-dimensional structure of mutant P53 visualized by the Swiss PDB Viewer.

Figure 2: Ramachandran plot of mutant P53 model obtained by PROCHECK: 88.6% residues in the favorable regions, 11.4% residues in
additional allowed regions, 0.0% residues in generously allowed regions and 0.0% in disallowed regions.

Figure 3: Mutant P53 protein structural validation using PROTSAV for Z plot (Black Spot) which describe the overall quality of model evaluated
and deciphered.

Figure 4: Mutant P53 protein structural validation using PROTSAV for verify 3D.

Figure 5: Mutant P53 protein structural validation using PROTSAV for quality verification plot of energy minimized model of the mutated p53
per-formed using ERRAT.

Figure 6: Mutant P53 protein structural validation using PROTSAV for Regions of the structure that can be rejected at the 95% confidence level
are yellow; regions that can be rejected at the 99% level are shown in red.
In addition to the previous qualitative assessments, the VADAR
statistics for quantitative evaluation of the predicted model
revealed that the model is structurally composed of alpha-helix
(17%), interspersed beta sheets (37%), coil (44%) and turn
(18%) with extensive H-bonding groups (Table 1) [25]. The Hbonds
distance and energy in the predicted secondary
structure protein were similar to the expected value, which
further corroborates the model’s good quality [26]. Likewise,
the calculation of the resolution of the protein from coordinate
data using ResProx showed a resolution of 1.8A (Table 2).
Generally, lower-resolution proteins below 2 A are highly
ordered, allowing individual hydrogen atoms to be visualized
and heavy atoms (C, O, N) to be very accurately mapped;
hence, they are a preferred choice for molecular docking.
The validation results, therefore, indicate that the refined
structure of the modelled protein is satisfactory and reliable for
subsequent computational studies [27]. The FindsiteCombo2.0
predicted the binding residues for mutated P53 [28]. These are
LEU 114, SER 116, THR 140, CYS 141, PRO 142, THR 102, ARG
110, LEU 111, TYR 126, ASN 131, ASN 268, SER 269, ASN 200,
GLU 221, PRO 222, GLU 224, THR 230, THR 231 and ILE 232.
Other binding pocket includes TYR 103, GLN 104, SER 366, ASN
310, SER 392, THR, 102, LYS 101, ASP 391, LYS 319, GLY 389, TRY
126, GLY 302, PHE 270, GLN 100, HIS 368, ASP 393, SER 314,
THR 312, GLU 271, ASN 311, ASP 352, THR 356, GLU 349, SER
269, PRO 128, LEU 330, GLU 326, ASP 268, ARG 110, GLN 144
and TYR 146.
| Protein structural details |
| Statistic |
Observed |
Expected |
| Helix |
42 (17%) |
- |
| Beta |
88 (37%) |
- |
| Coil |
104 (44%) |
- |
| Turn |
44 (18%) |
- |
| Hydrogen bonds |
| Mean hbond energy |
-1.8 sd=1.1 |
175 (75%) |
| Residue with hbonds |
159 (67%) |
Good: 0-1.5 |
| ResProx Result |
- |
Middle: 1.5-2.5 |
| Resolution of protein based on REPROX values = 1.874 ± 0.0 |
- |
Bad: > 2.5 |
Table 1: VADAR and ResProx results show the quantitative parameters of the modelled mutant P53 protein.
The expected values represent those numbers which could
be expected for highly refined X-ray and NMR protein
structures.
Molecular Docking Results
The docking simulations were carried out to discover potential
drug candidates to engage the active sites of the mutated p53 cellular tumor antigen in a way that will restore the anticancer
properties of the p53 gene, leading to therapeutic
measures in cancer research. To this effect, 50 compounds
were selected from 7 plants and were docked with 3Q05 protein. The selected ligand is covalently linked to the aimed
amino acid residues of the protein [29]. The reference
ligands’ (Gemcitabine and thiotepa) existence within the pocket is
subjected to ionic interactions and van der Waal forces with
different target residues comprising the p53 cellular tumor
antigen pocket. Consequently, gemcitabine and thiotepa were
considered the standard ligands to investigate the efficacy of the
selected phytochemicals to compete with it engaging the
mutated sites by activating a cancer responsive p53 gene [30].
Throughout the docking simulation process, the native ligands (Gemcitabine and thiotepa) were fitted in the binding sites of
the p53 cellular tumor antigen, forming conventional hydrogen
bonds with LYS120 and TYR126, respectively (binding
scores=-5.4 kcal/mol and -3.5 kcal.mol respectively).
The in silico docking of the target phytochemicals and the
standard ligand into the cellular tumor antigen protein were performed. Quite a number of receptor ligand positions were
gotten with better binding affinities and complex interactions
from the active receptor pockets. Poses with the most
qualitative RMSD values and higher ligand binding energies
were further analyzed [31]. Results of energies and receptor
ligand interactions with amino acids of the tumor antigen
pocket are presented in Table 2.
| List of plants |
Phytochemicals |
Docking score (Kcal/mol) |
Conf/Grid box dimension |
Binding sites |
| Morus alba |
Albanol B |
8.8 |
Receptor=3q05.pdbqt |
LEU 114, SER 116, THR 140, CYS 141, PRO 142, THR 102, ARG 110, LEU 111, TYR 126, ASN 131, ASN 268, SER 269, ASN 200, GLU 221, PRO 222, GLU 224, THR 230, THR 231 ILE 232 |
| Kuwanol A |
8.7 |
Exhaustiveness=8 |
| Morusin |
7.2 |
Center_x=-60.1681 |
| Aspalathus linearis |
Quercetin3-0-Robinobioside |
7.6 |
Size_z=25.0 |
| Thermopsoside |
7.1 |
Size_x=31.7390 |
| Size_y=32.2131 |
| Size_z=25.0 |
| Citrus paradisi |
Didymins |
7 |
Receptor=3q05.pdbqt |
TYR 103, GLN 104, SER 366, ASN 310, SER 392, THR, 102, LYS 101, ASP 391, LYS 319, GLY 389, TRY 126, GLY 302, PHE 270, GLN 100, HIS 368, ASP 393, SER 314, THR 312, GLU 271, ASN 311, ASP 352, THR 356, GLU 349, SER 269, PRO 128, LEU 330, GLU 326, ASP 268, ARG 110, GLN 144, TYR 146 |
| Hesparidin |
7.2 |
Exhaustiveness=8 |
| Naringin |
7.2 |
Center_x=-57.4290 |
| Narirutin |
7.6 |
Center_y=5.2404 |
| Neohesperidin |
7.3 |
Center_z=-8.4763 |
| Poncirin |
7.2 |
Size_x=41.1367 |
| Size_z=48.6496 |
| Campotheca acuminate |
Irinotecan |
8.9 |
Size_y=51.7491 |
- |
| Rubitecan |
7.1 |
Size_z=48.6496 |
- |
| 10-Hydroxycamptothecin |
7.5 |
- |
- |
| 9- Amino camptothecin |
7.2 |
- |
- |
| *Thiotepa |
3.5 |
- |
- |
| *Gemcitabine |
5.3 |
- |
- |
Table 2: Molecular docking scores of the phytochemicals and the standard drugs.
Most of the compounds came out with binding scores higher
than those of the ligands, but only those with binding energies
of -7.0 and lower were selected [32]. Out of the 15 selected
compounds, only three compounds showed the highest
binding affinities greater than -8.0 kcal/mol [33]. For Albanol,
binding interactions with 3Q05 (binding score=-8.8); three
conventional hydrogen bonds (ASN131, TYR126 and PHE131)
were observed, all of which are presumed to be important for
the process [34]. In addition, Pi-Anion, Pi-Akyl and Akyl
elements were noticed at these pockets ASP268, ARG110 and ARG110, respectively, whereas, in the case of Kuwanol,
binding interactions with 3Q05 (binding score=-8.7) and four
conventional hydrogen bonds also were noted, a Pi-donor
hydrogen bond was observed on the conventional hydrogen
bond site THR102 [35]. However, in the case of Irinotecan,
binding interactions with 3Q05 (binding score=-8.9), with one
conventional hydrogen identified (THR356) [36]. In addition, a
carbon-hydrogen bond exists with two pi-anions and an
alkyl (Figures 7-21).

Figure 7: 2D Interaction of Albanol B with active site of 3Q05.

Figure 8: 2D Interaction of Kuwanol A with active site of 3Q05.

Figure 9: 2D Interaction of thermopsoside with active site of 3Q05.

Figure 10: 2D Interaction of quercetin 3-O-robinobioside with active site of 3Q05.

Figure 11: 2D Interaction of morusin with active site of 3Q05.

Figure 12: 2D Interaction of didymin with active site of 3Q05.

Figure 13: 2D Interaction of hesperidin with active site of 3Q05.

Figure 14: 2D Interaction of naringin with the active site of 3Q05.

Figure 15: 2D Interaction of narirutin interaction with active site of 3Q05.

Figure 16: 2D Interaction of neohesperidin with active site of 3Q05.

Figure 17: 2D Interaction of poncirin with active site of 3Q05.

Figure 18: 2D Interaction of irinotecan with active site of 3Q05.

Figure 19: 2D interaction of rubitecan with active site of 3Q05.

Figure 20: 2D Interaction of 10-hydroxycamptothecin with active of 3Q05.

Figure 21: 2D interaction of 9-aminocamptothecin with active site of 3Q05.
Finally, other phytochemicals such as morusin, quercetin,
thermopsoside, didymins, hesperidin, naringin, nairutin,
rubitecan, 10-hydroxycamptothecin and 9-amino camptothecin
have outstanding binding energies (-7.2, -7.6, -7.1, -7.0, -7.2,
-7.2, -7.6, -7.3, -7.2, -7.1, -7.5 and -7.2, respectively), way better
than the standard ligands as shown in
Table 3. However, their
binding site was observed to have similar amino acid sequences
and positions. For morusin, a conventional hydrogen bond with
a Pi-anion and two pi-donor hydrogen bonds were observed
with GLU224 and THR231. Two conventional hydrogen
bonds were observed for quercetin at GLU224 and TYR229,
respectively. For 10-hydroxycamptothecin, one hydrogen bond was observed with SER269.
ADME Analysis Results
Lipinski’s rule of five was applied to estimate the drug likeliness
of all the selected 15 candidates with high binding affinity. This
evaluation technique aids in the elimination of a few chemicals
based on their physicochemical features. Compounds that
violated two or more characteristics were removed from the
list and the rest were considered for further analysis. Out of
the 15 ligands, 10 violated more than one parameter and five
compounds left were taken for toxicity test, as the results
shown in Table 3.
| S.N0 |
Compound name |
PubChem ID |
Compound structure |
Analysis |
|
| 1 |
Albanol B |
480819 |
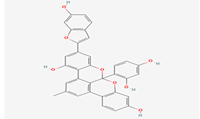 |
Molecular weight (<500 Da) |
558.53 |
| Lipophilicity (LogP <5) |
6.73 |
| H bond donor (<5) |
5 |
| H bond acceptor (<10) |
8 |
| Violations |
3 |
| 2 |
Kuwanol A |
14334319 |
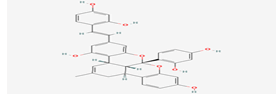 |
Molecular weight (<500 Da) |
564.58 |
| Lipophilicity (LogP <5) |
5.83 |
| H bond donor (<5) |
6 |
| H bond acceptor (<10) |
8 |
| Violations |
3 |
| 3 |
Thermopsoside |
11294177 |
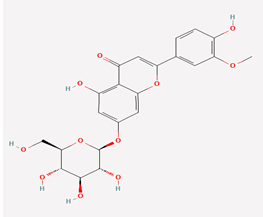 |
Molecular weight (<500 Da) |
462.40 1.79 |
| Lipophilicity (LogP <5) |
6 |
| H bond donor (<5) |
11 |
| H bond acceptor (<10) |
2 |
| Violations |
|
| 4 |
Quercetin 3-O-Robinobioside |
10371536 |
 |
Molecular weight (<500 Da) |
610.52 |
| Lipophilicity (LogP <5) |
-0.33 |
| H bond donor (<5) |
10 |
| H bond acceptor (<10) |
16 |
| Violations |
3 |
| 5 |
Morusin |
5281671 |
 |
Molecular weight (<500 Da) |
420.45 |
| Lipophilicity (LogP <5) |
5.52 |
| H bond donor (<5) |
3 |
| H bond acceptor (<10) |
6 |
| Violations |
1 |
| 6 |
Didymin |
16760075 |
 |
Molecular weight (<500 Da) |
594.56 |
| Lipophilicity (LogP <5) |
-0.66 |
| H bond donor (<5) |
7 |
| H bond acceptor (<10) |
14 |
| Violations |
3 |
| 7 |
Hesperidin |
10621 |
 |
Molecular weight (<500 Da) |
610.56 |
| Lipophilicity (LogP <5) |
-0.14 |
| H bond donor (<5) |
8 |
| H bond acceptor (<10) |
15 |
| Violations |
3 |
| 8 |
Naringin |
442428 |
 |
Molecular weight (<500 Da) |
580.53 |
| Lipophilicity (LogP <5) |
-0.44 |
| H bond donor (<5) |
8 |
| H bond acceptor (<10) |
14 |
| Violations |
2 |
| 9 |
Narirutin |
442431 |
 |
Molecular weight (<500 Da) |
580.53 |
| Lipophilicity (LogP <5) |
-0.99 |
| H bond donor (<5) |
8 |
| H bond acceptor (<10) |
14 |
| Violations |
3 |
| 10 |
Neohesperidin |
442439 |
 |
Molecular weight (<500 Da) |
610.56 |
| Lipophilicity (LogP <5) |
-0.47 |
| H bond donor (<5) |
8 |
| H bond acceptor (<10) |
15 |
| Violations |
3 |
| 11 |
Poncirin |
442456 |
 |
Molecular weight (<500 Da) |
594.56 |
| Lipophilicity (LogP <5) |
-0.11 |
| H bond donor (<5) |
7 |
| H bond acceptor (<10) |
14 |
| Violations |
3 |
| 12 |
Irinotecan |
60838 |
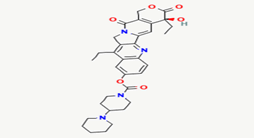 |
Molecular weight (<500 Da) |
586.68 |
| Lipophilicity (LogP <5) |
3.74 |
| H bond donor (<5) |
1 |
| H bond acceptor (<10) |
8 |
| Violations |
1 |
| 13 |
Rubitecan |
472335 |
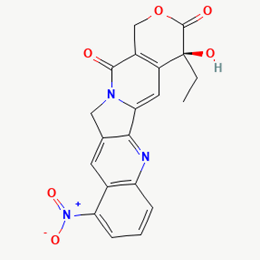 |
Molecular weight (<500 Da) |
393.35 |
| Lipophilicity (LogP <5) |
1.57 |
| H bond donor (<5) |
1 |
| H bond acceptor (<10) |
7 |
| Violations |
0 |
| 14 |
10-hydroxycamptothecin |
97226 |
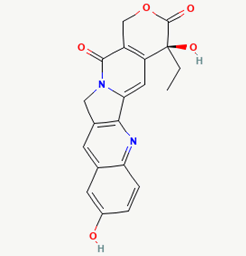 |
Molecular weight (<500 Da) |
364.35 |
| Lipophilicity (LogP <5) |
1.38 |
| H bond donor (<5) |
2 |
| H bond acceptor (<10) |
6 |
| Violations |
0 |
| 15 |
9-amino camptothecin |
|
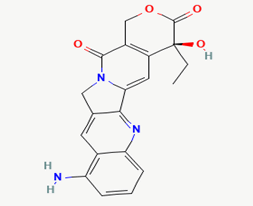 |
Molecular weight (<500 Da) |
363.37 |
| Lipophilicity (LogP <5) |
1.06 |
| H bond donor (<5) |
2 |
| H bond acceptor (<10) |
5 |
| Violations |
0 |
Table 3: ADMET Analysis of the fifteen phytochemicals.
Acute Toxicity Prediction
A substance’s acute toxic potential must be evaluated in
order to determine the adverse effects that may result from
accidental or intentional short-term exposure. The results of acute toxicity tests are used to guide dosage selection for longterm
toxicity studies and other animal-based studies (including
humans). The toxicity status of the test substance can be
determined based on the results of an acute toxicity test.
In silico toxicity of the selected lead compounds was predicted using the PASS online software. The majority of the leads were
observed to be in the QSAR models’ Applicability Domain (AD).
The toxicity predictions considered the IV, oral, subcutaneous and intraperitoneal routes, revealing that all of the
compounds belong to class 5. According to the analysis, the
compounds were safe and relatively harmless (Table 4).
| Compounds |
|
LD50 (mg/kg) |
|
|
| |
IP |
IV |
Oral |
SC |
| Morusin |
299,600a* |
151,200a* |
730,300a* |
361,900a* |
| Irinotecan |
166,700a* |
33,950a# |
236,500a# |
240,800a* |
| Rubitecan |
77,100b* |
101,500a* |
348,000b* |
199,000a* |
| 10-Hydroxycamptothecin |
47,170b# |
132,600a* |
1306,000b* |
196,400b* |
Note: IP, Intraperitoneal route; IV, Intravenous route; SC, Subcutaneous route; Oral; *Molecule falls in class 4; # Molecule falls in Class 3; a Compound falls in applicability domain of models; b Compound out of applicability domain of models.
Table 4: Acute toxicity prediction of lead compounds.
Density Functional Theory (DFT) Calculation
The stability of the hit compounds and their tendency to
undergo reactions required for the modulation of mutant P53
protein was estimated using the density functional theory.
The Frontier Molecular Orbitals (FMOs), which consist of
HOMO and LUMO give details information about the electron
donating and electron accepting capacity of a chemical
compound and their reactivity potential. HOMO energy
describes the electron donating capability of the compound,
while LUMO energy describes the electron accepting quality
of the compound. The difference between the HOMO and
LUMO values is denoted as ‘band gap’. The band gap
determines the stability of a compound; the larger the band
gap, the more stable compound and the smaller the value of
the energy gap of a compound, the more reactive and less
stable the compound. Irinotecan was observed to be the
most significant HOMO energy with values of -0.26342, which
aligns with the molecular docking score compared to the
standard drug, as shown in Table 5. The red and blue colour
indicates the positive and negative ± sign of the wave
function, respectively and the nodal nature of the orbital
HOMO and LUMO cloud were centralized majorly on the
aromatic rings [75], as shown in Table 6. According to the
energy gap of the compounds in Table 5, 10-
hydroxycamptothecin was observed as the most chemically
reactive and less stable compound with band energy of
-0.08953 eV, while Morusin was predicted as the least
chemically reactive and more stable compound with the band
gap of -0.16141 eV. Going by the FMOs calculated for the
reference compounds, Gemcitabine’s most reactive and less
stable compound. The parameters that heavily relied
on the HOMO and LUMO energies are given in Table 5. They are the global descriptors and they include Ionization Potential (IP), Electron Affinity (EA), chemical
hardness (η), chemical softness (ζ), electronegativity (χ),
chemical potential (μ) and electrophilicity (ω). They determine
the reactivity and stability of the compounds. Ionization
potential determines the chemical reactivity of molecules
high ionization potential denotes high stability and chemical
inertness and low ionization potential signifies low stability
but high reactivity. Electron affinity explains the electron
accepting capacity of a compound. Based on these, Irinotecan
is most stable and 10-hydroxycamptothecin is most reactive.
Electron affinity of the lead compound ranged between
-0.02946 eV and 0.13804 eV. Ionization Potential (IP) and
Electron Affinity (EA) reflect the redox stability of the drug, thus
easing metabolism. A recent study determined that clinically
approved drugs are well correlated with the experimental data
having EA values ranging between -1.5 and 2.0 eV. The E.A.
value for most of our lead compounds lies within this range,
corroborating their drug like potential. Using hardness and
softness as metrics to measure the reactivity and stability of
a chemical compound, the stability of a compound increases
with hardness but decreases with softness and the reactivity
of a compound decreases with hardness but increases with
softness. Based on this description, 10-hydroxycamptothecin
is a reactive compound. Overall, most of the hits compounds
had a significant reactivity to the reference compounds. The
electronegativity of a compound is the tendency of atoms in
a compound to attract a shared electron to itself. Irinotecan
is the most electronegative among the compounds. The hits
compound’s chemical potential (μ) was obtained as -0.119495
eV, -0.20073 eV, -0.172135 eV and -0.015305 eV, respectively.
The negative values for all the compounds imply good stability
and the formation of a stable complex with the receptor.
The electrophilicity index depicts information about electron
system structure, stability, bonding, reactivity and dynamics at
the ground and excited states. Irinotecan had the highest
electrophilicity index (0.321363), while the least electrophilicity index was observed in 10-hydroxycamptothecin (0.002616).
Compounds with a high electrophilicity index are more likely to
interact with biomolecules.
| Parameters |
Morusin |
Irinotecan |
Rubitecan |
10-hydroxycamptothecin |
Thiotepa |
Gemcitabine |
| HOMO |
-0.2002 |
-0.26342 |
-0.23266 |
-0.06007 |
-0.22579 |
0.03319 |
| LUMO |
-0.03879 |
-0.13804 |
-0.11161 |
0.02946 |
0.044 |
0.08591 |
| Band gap (eV) |
-0.16141 |
-0.12538 |
-0.12105 |
-0.08953 |
-0.26979 |
-0.05272 |
| Ionization potential (I) |
0.2002 |
0.26342 |
0.23266 |
0.06007 |
0.22579 |
-0.03319 |
| Electron affinity (A) |
0.03879 |
0.13804 |
0.11161 |
-0.02946 |
-0.044 |
-0.08591 |
| Hardness (ղ) |
0.080705 |
0.06269 |
0.060525 |
0.044765 |
0.134895 |
0.02636 |
| Softness (ζ) |
12.39081 |
15.95151 |
16.5221 |
22.33888 |
7.413173 |
37.93627 |
| Electronegativity (χ) |
0.119495 |
0.20073 |
0.172135 |
0.015305 |
0.090895 |
-0.05955 |
| Chemical potential (μ) |
-0.1195 |
-0.20073 |
-0.17214 |
-0.01531 |
-0.0909 |
0.05955 |
| Electrophilicity (ɷ) |
0.088465 |
0.321363 |
0.244779 |
0.002616 |
0.030623 |
0.067265 |
Table 5: Density Functional theory analysis of the lead compounds.
The Molecular Electrostatic Potential (MEP) of the compounds
was calculated to understand the distribution of charges over the
compound in a three dimensional format, thereby elucidating
the most electropositive (prone to nucleophilic attack) and
electronegative site (prone to electrophilic attack) [37-40]. The
MEP map of the compounds is illustrated. These sites provide
information on the atoms of the compounds that can form noncovalent
interactions. Different colours represent the values of
MEP at the surface of the compounds; red color signifies the
regions of the most electronegative electrostatic potential,
blue color shows the regions of the most electropositive electrostatic potential and grey colour represents the region
with zero potential. Therefore, the electrostatic potential
increases in the order red <grey <blue [41-43]. The
electrostatic potential contour map of the compounds, as
shown in Figures 22-27, demonstrated that the compounds
have many possible sites for electrophilic (red colour) and
nucleophilic attack (blue colour) [44-47]. The regions over the
compounds benzene ring are neutral as represented by grey
colour. It is important to note that these sites give details on
intermolecular interactions [48-50].

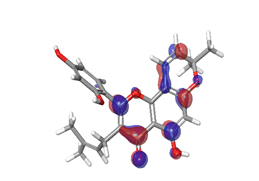
Figure 22: Irinotecan.

Figure 23: 10-hydroxycampotethecin.

Figure 24: Rubitecan.

Figure 25: Morusin.

Figure 26: Thiotepa.

Figure 27: Gemcitabine.
Discussion
Ovarian cancer is associated with late and advanced
diagnosis, which impedes prognosis and treatment due to
little to no effective screening methods [51-53]. The p53 gene
is implicated in most cancers, especially aggressive ovarian
cancer; this mutation is identified with the gene’s loss of
tumor suppressor capacity [54-57]. Different forms of
mutation are associated with the p53 gene, including
gene-level mutation, protein-level mutation, epigenetic
mutation or pure inactivation due to exploited pathways or
interactions e.g. the p53-MDM2 interaction [58-61].
Our study focuses on the mutation identified directly with the p53 gene, which leads to a loss of function of the wild-type p53 gene [62-65]. We tackled the loss of function of wild type
TP53 gene as related to ovarian cancer by modulating the activity of the mutant gene with phytochemicals. Charlotte
attributed the low survival rate of ovarian cancer patients to
the cost-demanding effect of diagnosis and treatment. This
phytochemical approach essentially aims to provide low-cost
treatment and increased survival of ovarian cancer patients
with reduced side effects [66-69]. The results of our study
show the modulating capacity of the novel phytochemicals
having passed all computational analysis and also shows
perfect imitation of the existing standards. Previous studies
have shown the use of lead compounds in the treatment of
other types of cancers [70-72]. Morusin, Irinotecan, Rubitecan
and camptothecin have been used to treat hepatocellular
carcinoma, colon cancer and BRCA-mutated platinumresistant
ovarian cancer (Table 6) [73].
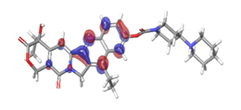
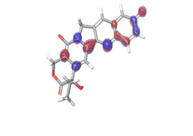
| HOMO |
Band gap |
LUMO |
A. Irinotecan 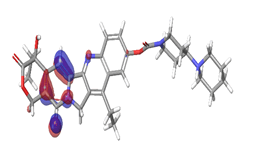 |
-0.12538 eV |
 |
| |
B. 10-Hydroxycamptothecin 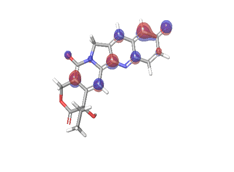 |
|
 |
| -0.08953eV |
| |
C. Rubitecan 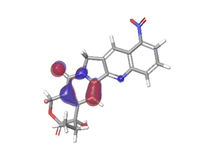 |
|
 |
| -0.12538eV |
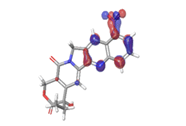
D. Morusin 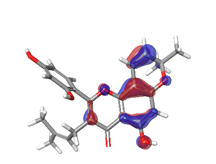 |
|
 |
| |
| |
| |
| |
| -0.16141eV |
E. Thiotepa 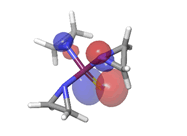 |
-0.26979eV |
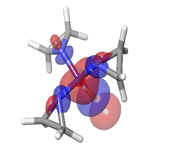 |
F. Gemcitabine 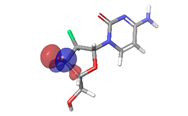 |
-0.05272eV |
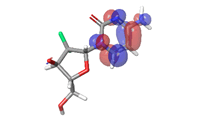 |
Table 6: HOMO and LUMO orbital structure of the lead compounds.
Conclusion
In summary, this study explored the therapeutic potential of
bioactive compounds from medicinal plants against mutant p53, a therapeutic target of ovarian cancer. Four bioactive
compounds were identified as the lead compounds (Irinotecan,
10-hydroxycamptothecin, Rubitecan and Morusin). These
compounds portrayed favourable binding with model mutant P53 than the reference compounds (standard inhibitors) and
interacted with essential amino acids that contribute to the
modulation of drug targets for better selectivity. In addition,
the lead compounds exhibit excellent pharmacokinetic
properties and less toxicity compared with the standard drugs.
DFT calculation revealed the atomic portion of the compounds
that either donate or accept electrons through HOMO, LUMO,
bandgap and global descriptors.
This study only considers a single type of mutant p53 amongst
a number of other mutants associated with ovarian cancer;
further studies are therefore encouraged to be done using
other identified mutants. We encourage a further wet lab
analysis on the phytochemicals suggested having passed all
computational analyses be carried out.
This study revealed four natural occurring bioactive compounds
found in medicinal plants as novel modulators of mutant P53. These compounds may present an explorable treatment regime
in the design of potent modulator mutant P53 in treating
ovarian cancer.
Ethics Approval and Consent to Participate
Not Applicable.
Consent for Publication
Not Applicable.
Availability of Data and Materials
All data and materials are available on request.
Competing Interests
The authors declare that they have no competing interests.
Funding
No external funding was received for this research.
Author’s Contributions
KYR, MAA, MMA, EJU, were involved in the design and
conceptualisation of the project, KYR, PFI, SAO , MA, KZB, MAA
and MIG contributed to project implementation, data analysis and manuscript writing. All authors have read and accepted
the manuscript.
Acknowledgements
The authors acknowledge the research support received
Omitogun Alamin Babatunde, Adeyemo Oluwatosin Maryam
and Dennis Peace Ezeobi.
References
- Guo T, Dong X, Xie S, Zhang L, Zeng P, et al. (2021) Cellular mechanism of gene mutations and potential therapeutic targets in ovarian cancer. Cancer Manag Res. 13:3081.
[Crossref] [Google Scholar] [PubMed]
- Lee DF, Su J, Kim HS, Chang B, Papatsenko D, et al. (2015) Modeling familial cancer with induced pluripotent stem cells. Cell. 161:240-254.
[Crossref] [Google Scholar] [PubMed]
- Lisio MA, Fu L, Goyeneche A, Gao ZH, Telleria C (2019) High grade serous ovarian cancer: Basic sciences, clinical and therapeutic standpoints. Int J Mol Sci. 20:952.
[Crossref] [Google Scholar] [PubMed]
- Salehi F, Dunfield L, Phillips KP, Krewski D, Vanderhyden BC (2008) Risk factors for ovarian cancer: An overview with emphasis on hormonal factors. J Toxicol Environ Health, Part B. 11:301-321.
[Crossref] [Google Scholar] [PubMed]
- Ledermann JA, Raja FA, Fotopoulou C, Gonzalez-Martin A, Colombo N, et al. (2013) Newly diagnosed and relapsed epithelial ovarian carcinoma: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Annal Oncol. 24:24-32.
[Crossref] [Google Scholar] [PubMed]
- Rivlin N, Brosh R, Oren M, Rotter V(2011) Mutations in the p53 tumor suppressor gene: Important milestones at the various steps of tumorigenesis. Genes Cancer. 2:466-474.
[Crossref] [Google Scholar]
- Duffy MJ, Synnott NC, O’Grady S, Crown J (2022) Targeting p53 for the treatment of cancer. Semin Cancer Biol. 79: 58-67.
[Crossref] [Google Scholar] [PubMed]
- Levine AJ (1997) p53, the cellular gatekeeper for growth and division. cell. 88:323-331.
[Crossref] [Googlescholar] [PubMed]
- Zilfou JT, Lowe SW (2009) Tumor suppressive functions of p53. Cold Spring Harb Perspect Biol. 1:a001883.
[Crossref] [Google Scholar] [PubMed]
- Menichini P, Monti P, Speciale A, Cutrona G, Matis S, et al. (2021) Antitumor effects of PRIMA-1 and PRIMA-1 Met (APR246) in hematological malignancies: Still a mutant P53-dependent affair. Cells. 10:98.
[Crossref] [Google Scholar] [PubMed]
- Perdrix A, Najem A, Saussez S, Awada A, Journe F, et al. (2017) PRIMA-1 and PRIMA-1Met (APR-246): From mutant/wild type p53 reactivation to unexpected mechanisms underlying their potent anti-tumor effect in combinatorial therapies. Cancers. 9:172.
[Crossref] [Google Scholar] [PubMed]
- Chen J (2016) The cell cycle arrest and apoptotic functions of p53 in tumor initiation and progression. Cold Spring Harb Perspect Biol. 6: 026104.
[Crossref] [Googlescholar] [PubMed]
- Klymkowsky MW, Savagner P (2009) Epithelial mesenchymal transition: A cancer researcher's conceptual friend and foe. Am J Pathol. 174:1588-1593.
[Crossref] [Google Scholar] [PubMed]
- Omar SI, Tuszynski J (2018) The molecular mechanism of action of methylene quinuclidinone and its effects on the structure of p53 mutants. Oncotarget. 9:37137.
[Crossref] [Google Scholar] [PubMed]
- Kaur RP, Vasudeva K, Kumar R, Munshi A (2018) Role of p53 gene in breast cancer: Focus on mutation spectrum and therapeutic strategies. Curr Pharm Des. 24:3566-3575.
[Crossref] [Google Scholar] [PubMed]
- Stiewe T, Haran TE (2018) How mutations shape p53 interactions with the genome to promote tumorigenesis and drug resistance. Drug Resist Updat. 38:27-43.
[Crossref] [Google Scholar] [PubMed]
- Freed-Pastor WA, Prives C (2012) Mutant p53: One name, many proteins. Genes Dev. 26:1268-1286.
[Crossref] [Google Scholar] [PubMed]
- Cole AJ, Dwight T, Gill AJ, Dickson KA, Zhu Y, et al. (2016) Assessing mutant p53 in primary high-grade serous ovarian cancer using immunohistochemistry and massively parallel sequencing. Sci Rep. 6:1-2.
[Crossref] [Google Scholar] [PubMed]
- Kupryjanczyk J, Thor AD, Beauchamp R, Merritt V, Edgerton SM, et al. (1993) P53 gene mutations and protein accumulation in human ovarian cancer. Proc Natl Acad Sci. 90:4961-4965.
[Crossref] [Google Scholar] [PubMed]
- Mazars R, Pujol P, Maudelonde T, Jeanteur P, Theillet (1991) P53 mutations in ovarian cancer: A late event. Oncogene. 6:1685-1690.
[Google Scholar] [PubMed]
- Oda K, Ikeda Y, Kashiyama T, Miyasaka A, Inaba K, et al. (2016) Characterization of TP53 and PI3K signaling pathways as molecular targets in gynecologic malignancies. J Obstet Gynaecol Res. 42:757-762.
[Crossref] [Google Scholar] [PubMed]
- Zanjirband M, Edmondson RJ, Lunec J (2016) Pre-clinical efficacy and synergistic potential of the MDM2-p53 antagonists, Nutlin-3 and RG7388, as single agents and in combined treatment with cisplatin in ovarian cancer. Oncotarget. 7:40115.
[Crossref] [Google Scholar] [PubMed]
- Amin AR, Karpowicz PA, Carey TE, Arbiser J, Nahta R, et al. (2015) Evasion Of Anti-Growth Signaling: A Key Step In Tumorigenesis And Potential Target For Treatment And Prophylaxis By Natural Compounds. Inseminars Cancer Biol. 35:55-77.
[Crossref] [Google Scholar] [PubMed]
- Li WW, Johnson-Ajinwo OR, Uche FI (2016) Advances of plant-derived natural products in ovarian cancer therapy. Int J Canc Prev. 9:181-185.
[Google Scholar]
- Yang X, Yang L, Zheng H (2010) Hypolipidemic and antioxidant effects of mulberry (Morus alba L.) fruit in hyperlipidaemia rats. Food Chem Toxicol. 48:2374-2379.
[Crossref] [Googlescholar] [PubMed]
- Kim JK, Kim M, Cho SG, Kim MK, Kim SW, et al. (2010) Biotransformation of mulberroside a from Morus alba results in enhancement of tyrosinase inhibition. J Ind Microbiol Biotechnol. 37:631-637.
[Crossref] [Google Scholar] [PubMed]
- Kuete V, Fozing DC, Kapche WF, Mbaveng AT, Kuiate JR, et al. (2009) Antimicrobial activity of the methanolic extract and compounds from Morus mesozygia stem bark. J Ethnopharmacol. 124:551-555.
[Crossref] [Google Scholar] [PubMed]
- Soonthornsit N, Pitaksutheepong C, Hemstapat W, Utaisincharoen P, Pitaksuteepong T (2017) In vitro anti-inflammatory activity of Morus alba L. stem extract in LPS-stimulated raw 264.7 cells. Evid Based Complement Altern Med.
[Crossref] [Google Scholar] [PubMed]
- Yiemwattana I, Chaisomboon N, Jamdee K (2018) Antibacterial and anti-inflammatory potential of Morus alba stem extract. Open Dent J. 12:265.
[Crossref] [Google Scholar] [PubMed]
- Jiao Y, Wang X, Jiang X, Kong F, Wang S, et al. (2017) Antidiabetic effects of Morus alba fruit polysaccharides on high-fat diet-and streptozotocin-induced type 2 diabetes in rats. J Ethnopharmacol. 199:119-127.
[Crossref] [Google Scholar] [PubMed]
- Eo HJ, Park JH, Park GH, Lee MH, Lee JR, et al. (2014) Anti-inflammatory and anti-cancer activity of mulberry (Morus alba L.) root bark. BMC Complement Altern Med. 14:1-9.
[Crossref] [Google Scholar] [PubMed]
- Saranya J, Shilpa G, Raghu KG, Priya S (2017) Morus alba Leaf Lectin (MLL) sensitizes MCF-7 cells to anoikis by inhibiting fibronectin mediated integrin-FAK signaling through RAS and activation of P38 MAPK. Front Pharmacol. 8:34.
[Crossref] [Google Scholar] [PubMed]
- Liu LF, Desai SD, LI TK, Mao Y, Sun ME, et al. (2000) Mechanism of action of camptothecin. Ann Acad Sci. 922:1-0.
[Crossref] [Google Scholar] [Indexed]
- Dong Q, Luo J, Qiu W, Cai L, Anjum SI, et al. (2016) Inhibitory effect of camptothecin against rice bacterial brown stripe pathogen Acidovorax avenae subsp. Avenae RS-2 Mol. 21:978.
[Crossref] [Google Scholar] [PubMed]
- Domalaon R, Ammeter D, Brizuela M, Gorityala BK, Zhanel GG, et al. (2019) Repurposed antimicrobial combination therapy: Tobramycin-ciprofloxacin hybrid augments activity of the anticancer drug mitomycin C against multidrug-resistant gram-negative bacteria. Front Microbiol. 10:1556.
[Crossref] [Google Scholar] [PubMed]
- Partridge FA, Poulton BC, Lake MA, Lees RA, Mann HJ, et al. (2021) Actions of camptothecin derivatives on larvae and adults of the arboviral vector Aedes aegypti. Molecules. 26:6226.
[Crossref] [Google Scholar] [PubMed]
- Yin D, Yin L, Wang J, Shen X, Dai Y, et al. (2022) Antiviral and virucidal activities of camptothecin on fowl adenovirus serotype 4 by blocking virus replication front cell. Infect Microbiol. 442.
[Crossref] [Google Scholar] [PubMed]
- Rabe C, Steenkamp JA, Joubert E, Burger JF, Ferreira D (1994) Phenolic metabolites from rooibos tea (Aspalathus linearis). Phytochem. 35:1559-1565.
[Crossref] [Google Scholar]
- Bramati L, Minoggio M, Gardana C, Simonetti P, Mauri P, et al. (2002) Quantitative characterization of flavonoid compounds in rooibos tea (Aspalathus linearis) by LC-UV/DAD. J Agric Food Chemistry. 50:5513-5519.
[Crossref] [Google Scholar] [PubMed]
- Shimamura N, Miyase T, Umehara K, Warashina T, Fujii S (2006) Phytoestrogens from Aspalathus linearis. Biol Pharm Bull. 29:1271-1274.
[Crossref] [Google Scholar] [PubMed]
- Krafczyk N, Glomb MA (2008) Characterization of phenolic compounds in rooibos tea. J Agric Food Chem. 56:3368-3376.
[Crossref] [Google Scholar] [PubMed]
- Srivastava S, Somasagara RR, Hegde M, Nishana M, Tadi SK, et al. (2016) Quercetin, a natural flavonoid interacts with DNA, Arrests cell cycle and causes tumor regression by activating mitochondrial pathway of apoptosis. Sci Rep. 6:1-3.
[Crossref][Google Scholar] [PubMed]
- Xi L, Zhang Y, Kong S, Liang W (2018) MIR-34 inhibits growth and promotes apoptosis of osteosarcoma in nude mice through targetly regulating tgif2 expression. Biosci Rep. 38(3).
[Crossref] [Google Scholar] [PubMed]
- Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, et al. (2000) The protein data bank. Nucleic Acids Res. 28:235-242.
[Crossref] [Google Scholar] [PubMed]
- Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, et al. (1997) Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25:3389-3402.
[Google Scholar]
- Petty TJ, Emamzadah S, Costantino L, Petkova I, Stavridi ES, et al. (2011) An induced fit mechanism regulates p53 DNA binding kinetics to confer sequence specificity. EMBO J. 30:2167-2176.
[Crossref] [Google Scholar] [PubMed]
- Zhou H, Cao H, Skolnick J (2018) FINDSITEcomb2. 0: A new approach for virtual ligand screening of proteins and virtual target screening of biomolecules. J Chem Inf Model. 58:2343-2354.
[Crossref] [Google Scholar] [PubMed]
- Schwede T, Kopp J, Guex N, Peitsch MC (2003) SWISS-MODEL: An automated protein homology-modeling server. Nucleic Acids Res. 31:3381-3385.
[Crossref] [Google Scholar] [PubMed]
- Arnold K, Bordoli L, Kopp J, Schwede T (2006) The SWISS-MODEL workspace: A web-based environment for protein structure homology modelling. Bioinform. 22:195-201.
[Crossref] [Google Scholar] [PubMed]
- Laskowski RA, MacArthur MW, Moss DS, Thornton JM (1993) Procheck: A program to check the stereochemical quality of protein structures. J Appl Crystallogr. 26:283-291.
[Crossref] [Google Scholar]
- Colovos C, Yeates TO (1993) Verification of protein structures: Patterns of nonbonded atomic interactions. Protein Sci. 2:1511-1519.
[Crossref] [Google Scholar] [PubMed]
- Bowie JU, Lüthy R, Eisenberg D (1991) A method to identify protein sequences that fold into a known three-dimensional structure. Sci. 253:164-170.
[Crossref] [Google Scholar] [PubMed]
- Berjanskii M, Zhou J, Liang Y, Lin G, Wishart DS (2012) Resolution-by-proxy: A simple measure for assessing and comparing the overall quality of NMR protein structures. J Biomol NMR. 53:167-180.
[Crossref] [Google Scholar] [PubMed]
- Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, et al. (2009) AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J Comput Chem. 30:2785-2791.
[Crossref] [Google Scholar] [PubMed]
- Kim S, Chen J, Cheng T, Gindulyte A, He J, et al. PubChem in 2021: New data content and improved web interfaces. Nucleic Acids Res. 49:1388-1395.
[Crossref] [Google Scholar]
- Eberhardt J, Santos-Martins D, Tillack AF, Forli S (2021) AutoDock Vina 1.2.0: New docking methods, expanded force field and python bindings. J Chem Inform Model. 61:3891-3898.
[Crossref] [Google Scholar] [PubMed]
- Korobko D (2016) Synthesis of the row of new functional derivatives of 7-arylalkyl-8-hydrazine theophyllines. Science Rise. 3:39-45.
[Crossref] [Google Scholar]
- Alyar S, Sen T, Özmen ÜÖ, Alyar H, Adem S, et al. (2019) Synthesis, spectroscopic characterizations, enzyme inhibition, molecular docking study and DFT calculations of new Schiff bases of sulfa drugs. J Mol Struct. 1185:416-424.
[Crossref] [Google Scholar]
- Dalton JA, Jackson RM (2007) An evaluation of automated homology modelling methods at low target-template sequence similarity. Bioinform. 23:1901-1908.
[Crossref] [Google Scholar] [PubMed]
- Clemedson C, McFarlane-Abdulla E, Andersson M, Barile FA, Calleja MC, et al. (1996) MEIC evaluation of acute systemic toxicity: Part II. In vitro results from 68 toxicity assays used to test the first 30 reference chemicals and a comparative cytotoxicity analysis. Altern Lab Anim. 24(1_part_1):273-311.
[Crossref] [Google Scholar]
- Abraham CS, Muthu S, Prasana JC, Armakovic S, Armakovic SJ, et al. (2019) Computational evaluation of the reactivity and pharmaceutical potential of an organic amine: A DFT, molecular dynamics simulations and molecular docking approach. Spectrochim Acta A Mol Biomol Spectrosc. 222:117188.
[Crossref] [Google Scholar] [PubMed]
- Fang G, Xu L, Cao Y, Li A (2016) Theoretical design and computational screening of precursors for atomic layer deposition. Coord Chem Rev. 322:94-103.
[Crossref] [Google Scholar]
- Olawale F, Olofinsan K, Iwaloye O, Ologuntere TE (2021) Phytochemicals from Nigerian medicinal plants modulate therapeutically-relevant diabetes targets: Insight from computational direction. Adv Tradit Med. 1-5.
[Google Scholar]
- Olawale F, Iwaloye O, Olofinsan K, Ogunyemi OM, Gyebi GA, et al. (2022) Homology modelling, vHTS, pharmacophore, molecular docking and molecular dynamics studies for the identification of natural compound-derived inhibitor of MRP3 in acute leukaemia treatment. Chem Pap. 76:3729-3757.
[Google Scholar]
- Kausar T, Nayeem SM (2018) Identification of small molecule inhibitors of ALK2: A virtual screening, density functional theory and molecular dynamics simulations study. J Mol Model. 24:1-5.
[Crossref] [Google Scholar] [PubMed]
- Matuszek AM, Reynisson J (2016) Defining known drug space using DFT. Mol Inform. 35:46-53.
[Crossref] [Google Scholar] [PubMed]
- Asati V, Thakur SS, Upmanyu N, Bharti SK (2018) Virtual screening, molecular docking and DFT studies of some Thiazolidine-2, 4-diones as potential PIM-1 Kinase inhibitors. Chem Select. 3:127-135.
[Google Scholar]
- Ganesan MS, Raja KK, Murugesan S, Kumar BK, Rajagopal G, et al. (2020) Synthesis, biological evaluation, molecular docking, molecular dynamics and DFT studies of quinoline-fluoroproline amide hybrids. J Mol Struct. 1217:128360.
[Crossref] [Google Scholar]
- Ramya N, Jagadeeswari P, BIST B (2017) Proper coloring of regular graphs. Int J Pure Appl Math. 116.
[Google Scholar]
- Chinnasamy S, Selvaraj G, Kaushik AC, Kaliamurthi S, Nangraj AS, et al. Identification of potent inhibitors against aurora kinase A using molecular docking and molecular dynamics simulation studies.
[Google Scholar]
- Sfakianos GP, Havrilesky LJ (2011) A review of cost-effectiveness studies in ovarian cancer. Cancer Control. 18(1):59-64.
[Crossref] [Google Scholar] [PubMed]
- Marcus CS, Maxwell GL, Darcy KM, Hamilton CA, McGuire WP (2014) Current approaches and challenges in managing and monitoring treatment response in ovarian cancer. J Cancer. 5:25-30.
[Crossref] [Google Scholar] [PubMed]
- Wan LZ, Ma B, Zhang YQ (2014) Preparation of morusin from Ramulus mori and its effects on mice with transplanted H22 hepatocarcinoma. Biofactors. 40:636-645.
[Crossref] [Google Scholar] [PubMed]
- Gao L, Wang L, Sun Z, Li H, Wang Q, et al. Morusin shows potent antitumor activity for human hepatocellular carcinoma in vitro and in vivo through apoptosis induction and angiogenesis inhibition. Drug Des Devel Ther. 11:1789.
[Crossref] [Google Scholar] [PubMed]
- Huang M, Liu C, Shao Y, Zhou S, Hu G, et al. (2022) Anti-tumor pharmacology of natural products targeting mitosis. Cancer Biol Med. 19:774.
[Crossref] [Google Scholar] [PubMed]
Citation: Raheem K (2023) Therapeutic Capability of Selected Medicinal Plants Bioactive Constituents Against the Mutant Ovarian TP53 Gene; A Computational Approach. Res J Onco. 7:12.
Copyright: © 2023 Raheem K. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.