Keywords
Pancreatic Diseases; Pancreatic Neoplasms; Pancreaticoduodenectomy
Abbreviations
DP: distal pancreatectomy; FAP: familial adenomatous polyposis; NSE: neuron-specific enolase; PPPD: pyloruspreserving pancreaticoduodenectomy; SPT: solid pseudopapillary tumour
Note
Case 5 and its accompanying Figure 6 were added after the peer-review process. This case reinforces the theme of the paper and in no way alters the discussion and concluding remarks.
INTRODUCTION
Solid pseudopapillary tumour (SPT) of the pancreas is an extremely rare epithelial tumour of low malignant potential affecting predominantly adolescent girls and young women. It was first described by Frantz in 1959 [1] and has since then been referred to by a variety of terms, including Frantz’s tumour, solid-cystic tumour, papillary-cystic tumour, solid and papillary epithelial neoplasm and solid pseudopapillary tumour of the pancreas. The descriptive nature of the nomenclature reflects the uncertainty over the histogenesis of these tumours, which remains despite a growing body of literature. SPTs are characterized by unique clinical and pathological features. They are most common in females (90%) and occur predominantly at a young age (mean age 35 years with less than 10% over 40 years) [2, 3]. They are more frequent in non-Caucasians with the highest incidence in Japan [3]. They are regarded as slow-growing relatively benign tumours, though metastasis has been reported. We report five cases which demonstrate the remarkable variability of SPTs in their clinical presentation, histological and immunohistochemical features and biological behaviour.
CASE REPORTS
Case 1
A 34-year-old Caucasian lady presented acutely with epigastric pain of 24-hour duration, a two week history of jaundice and 3.5 kg of weight loss. Laboratory findings were normal apart from a raised alkaline phosphatase (1,560 IU/L; reference range: 70- 300 IU/L) and total bilirubin (50 μmol/L; reference range: 5-21 μmol/L). Tumour markers (alpha-fetoprotein, CA 125 and CA 19.9) were normal. Abdominal examination was unremarkable. Ultrasound (US) examination showed an echogenic mass in the pancreatic head with intra- and extra-hepatic duct dilatation. On computed tomography (CT) the mass measured 11cm in maximum diameter, and was shown to displace but not infiltrate adjacent vascular structures. There was no evidence of metastasis. Laparotomy revealed a large tumour mass arising from the pancreatic head with the duodenum stretched over its surface. Pyloruspreserving pancreaticoduodenectomy (PPPD) was performed. The tumour size was 11x9.5x6.5 cm. Histologically, most parts of the tumour showed features characteristic of SPT. In addition, however, there were areas of diffuse, densely cellular tumour growth, with marked nuclear atypia, prominent mitotic activity, and extensive necrosis (Figure 1a). This atypical tumour component also showed an aberrant immunoprofile, with acquisition of epithelial (MNF116, CAM5.2, BerEP4, EMA), endocrine (synaptophysin) and melanocytic (HMB45) marker expression (Figure 1b; Table 1). There was lymphovascular tumour permeation and metastatic spread to 7 out of 28 lymph nodes. Adjuvant treatment was offered but the patient declined. Post-operative recovery was uneventful and the patient was discharged with a follow-up appointment after two months. However, nearly two months later she presented acutely with abdominal pain and general deterioration. CT then showed liver metastases and she died two weeks later.
Figure 1. a. Atypical component in aggressive SPT
(Case 1): solid tumour sheets with little intervening
stroma show extensive necrosis (upper right corner),
severe nuclear atypia and multiple mitotic figures
(arrows; H&E stain x200). b. The atypical tumour cells
show aberrant expression of HBM45, a melanocytic
marker (peroxidase immunostain x400).
Case 2
A 64-year-old Sudanese lady was found to have a right upper abdominal mass on routine examination. She was asymptomatic. Laboratory findings were all normal. US examination showed a 9 cm solid mass, which on CT appeared to originate from the uncinate process. US-guided biopsy was suggestive of a neuroendocrine tumour (Figure 2). Magnetic resonance imaging (MRI) showed a mass with high signal intensity on T2 and a relatively low signal on T1 (Figure 3). Exploration revealed a huge hypervascular tumour arising from the head of the pancreas. A PPPD was performed. The tumour was well-circumscribed and measured 12.5x9.5x6.5 cm. The tumour showed diffuse haemorrhage and a central area of necrosis with cavity formation (Figure 4). Histology was consistent with SPT with no lymph node metastasis (Figure 5). Immunohistochemistry was unusual in that the majority of tumour cells stained for the endocrine marker synaptophysin (Table 1). The patient is well at 18-month follow-up.
Figure 2. US-guided biopsy in Case 2. Solid growth
pattern, minimal cytological atypia (a.) and
immunostaining for synaptophysin (b.) resulted in an
erroneous pre-operative diagnosis of a neuroendocrine
tumour (residual pancreas in lower part of micrograph;
x200).
Figure 3. MRI with gadolinium enhancement in Case
2. Tumour in the head of the pancreas (A). Portal vein
(B).
Figure 4. Cross-section of a large SPT (Case 2) with
extensive haemorrhage and necrosis.
Figure 5. Microscopic appearance of SPT (Case 2):
characteristic pseudopapillary growth pattern with gaplike
spaces and delicate fibrovascular stalks lined by a
single row of tumour cells (H&E stain x100).
Case 3
A 12-year-old Caucasian boy presented acutely after falling over and landing on a hockey ball which he had been carrying in his hands. He complained of epigastric pain and vomiting. A mass was palpable in the left upper abdomen. All laboratory findings were normal apart from an elevated total bilirubin (31 μmol/L) and amylase (129 IU/L; reference range: 0-110 IU/L); serum beta- HCG and alpha-fetoprotein were normal. Haemaglobin fell from 12.5 to 11.5 g/dL (reference range: 13.5-18.0 g/dL) within 24 hours of admission. US and CT examination showed a large, well-circumscribed round mass in intimate relationship to the body and tail of the pancreas, suggestive of a pancreatic tumour possibly with internal haemorrhage (Figure 6).
Figure 6. Abdominal CT in Case 3 showing a large,
well-circumscribed round mass in intimate relationship
to the body and tail of the pancreas, suggestive of a
pancreatic tumour possibly with internal haemorrhage.
At laparotomy, there was a large, tense, and solid, partly cystic tumour in the body of the pancreas. There was evidence of retroperitoneal rupture of the tumour with haemorrhage into the transverse mesocolon and retroperitoneum around the duodenum. The liver appeared normal and there was no intra-abdominal lymphadenopathy. There was evidence of portal hypertension with dilated mesenteric and splenic veins related to compression by the tumour. Complete resection was achieved by a distal pancreatectomy (DP). The tumour measured 7x7x4 cm and at the site of rupture of the tumour capsule, there were signs of haemorrhage but no extracapsular tumour growth. Histology was consistent with SPT. Post-operative recovery was uneventful and at six years he remains disease free.
Case 4
A 28-year-old Caucasian lady presented with a 24-hour history of epigastric and right upper abdominal pain associated with vomiting and fever. Laboratory findings were normal apart from an elevated CRP (62 mg/L; reference range: 0-10.0 mg/L). US revealed a mass in the head of the pancreas and CT confirmed a 4 cm mixed attenuation lesion of either neoplastic or inflammatory nature. CT repeated six weeks later showed a 3.5 cm cystic lesion without signs of inflammation. Subsequent MRI demonstrated a well-defined spherical mass within the pancreatic head which on endoscopic US was shown to have solid and cystic components. The acute presentation was assumed to be precipitated by a bleed into the tumour, while the observed decrease in size was thought to reflect resolution of the haematoma.
Laparotomy revealed a well-circumscribed tumour in the pancreatic head and neck which abutted onto but did not involve the portal vein. Complete resection was achieved by PPPD. The tumour was partially encapsulated and measured 2.2x1.8x1.4 cm. Histology confirmed SPT. There was focal perineural tumour permeation but no lymph node metastasis. Post-operative recovery was uneventful and at five-year follow-up the patient remains disease free.
Case 5
A 10-year-old Asian girl was referred with a one year history of mild, fluctuating jaundice and intermittent central abdominal pain. She had been born to consanguineous parents and had required treatment for unconjugated hyperbilirubinaemia in the neonatal period. During an admission for a febrile convulsion at 18 months of age her total bilirubin was 54 μmol/L. Examination revealed no abnormal physical signs apart from mild jaundice. Investigations included the following: an elevated plasma total bilirubin of 84 μmol/L (conjugated fraction 4 μmol/L) but otherwise normal biochemical liver function, normal full blood count, reticulocyte count, and blood film. She was considered to have either Crigler-Najjar type 2 or Gilbert disease as a cause of her unconjugated hyperbilirubinemia. An abdominal ultrasound scan revealed a 5.5 cm well-defined heterogeneous retroperitoneal mass in the region of the head of the pancreas displacing but not obstructing the common bile duct. Colour Doppler flow studies showed the mass to be relatively avascular. Abdominal CT confirmed a well-demarcated mass related to the head of the pancreas immediately anterior to the inferior vena cava. The pancreas appeared otherwise normal and there was no associated lymphadenopathy. Other investigations included a normal plasma amylase, serum human chorionic gonadotrophin, alpha-fetoprotein, lactate dehydrogenase, and chest radiograph.
At laparotomy, she was found to have a large pancreatic head tumour which was firm, encapsulated, and not associated with evidence of local invasion or intra-abdominal metastases. She underwent a PPPD with complete macroscopic excision of the tumour. She made an uneventful postoperative recovery and four years later remains well with normal pancreatic function and no evidence of recurrent disease.
Immunohistochemistry
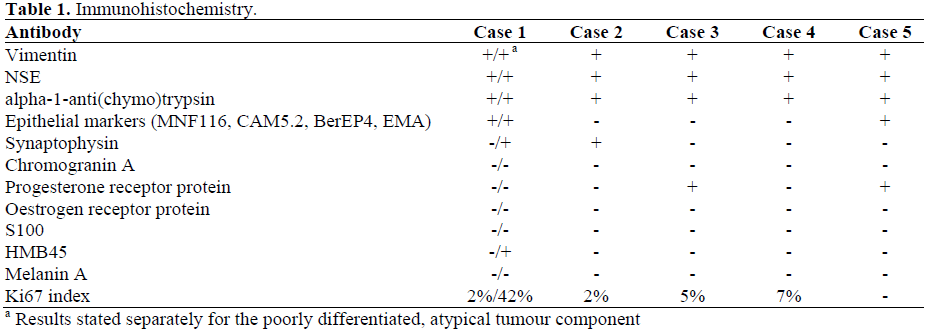
The summary of immunohistochemistry for all five tumours is presented in Table 1.
DISCUSSION
SPTs are uncommon, accounting for 1-2% of all exocrine pancreatic tumours [2]. They present mainly in the second and third decade of life, being rare in the first decade and occurring in less than 10% above the age of 40 years [3]. SPTs are often found incidentally on routine examination in an asymptomatic individual or as an incidental finding. Other patients may present with abdominal pain, palpable mass, dyspepsia, bloating or obstructive jaundice [4]. There may be no abnormal laboratory findings, and typically serum amylase and tumour markers (CA 19.9, CA 125, CEA, and alpha-fetoprotein) are normal.
US, CT and MRI have all been used to investigate these masses. US and CT usually show a well-demarcated multilocular cystic mass with internal septa, fluid-filled areas and solid portions [5, 6]. MRI may demonstrate haemorrhage into the tumour [7]. None of these appearances are diagnostic and need to be considered in the clinical context.
Macroscopically, SPTs are usually large tumours at the time of presentation (mean diameter 10.3 cm), often with extensive haemorrhage and pseudocystic degeneration [3]. SPTs are well-demarcated, often surrounded by an (incomplete) capsule, and only rarely do they invade adjacent organs. They are usually solitary tumours which are evenly distributed throughout the pancreas [3], though unusual multicentric [8] or extrapancreatic (mesocolonic, retroperitoneal, omental, hepatic) [9, 10] presentations have been reported.
Histologically, most SPTs show a solidmonomorphous growth in peripheral parts, whereas in the centre, tumour cells become dyscohesive and form pseudopapillae surrounded by empty gap-like spaces. Branching papillary fronds, cytological monotony and nuclear grooving are characteristic features that allow confident cytological diagnosis [11]. Degenerative changes feature prominently and lead not only to tumour cell vacuolization and the formation of characteristic pseudopapillae, but can also result in extensive fibrosis with focal calcification and occasional ossification, collections of foamy macrophages, and haemorrhage with cholesterol clefts and foreign body giant cell reaction. There is usually a delicate and endocrine-like tumour vasculature.
SPTs need to be differentiated from endocrine tumours, which morphologically can be very similar, but have a poorer prognosis. While endocrine tumours usually lack the widespread degenerative features, pseudopapillary growth and nuclear grooving of SPTs, immunohistochemistry is invaluable to solve the differential diagnosis. SPTs exhibit diffuse strong labelling for vimentin and neuron-specific enolase (NSE), whereas immunostaining for epithelial markers is usually absent. Staining for alpha-1- antitrypsin and alpha-1-antichymotrypsin shows a characteristic pattern of strongly positive tumour cells scattered as singletons or small clusters within a background of negative tumour cells. Positivity for chromogranin A and synaptophysin has been reported in some SPTs and underscores the importance of a broad panel of markers upon which to base the diagnosis. In Case 2, positive immunostaining for synaptophysin in solid parts of the tumour sampled by USguided biopsy had resulted in an erroneous pre-operative diagnosis of neuro-endocrine tumour. Many SPTs express progesterone receptors, while oestrogen receptors are usually absent. Other differentials to consider are acinar cell carcinoma, and in children under 10 years of age, pancreatoblastoma. Acinar cell carcinoma has a male sex predilection, lobulated and ill-demarcated gross appearance, lack of pseudopapillary changes and positive immunostaining for pancreatic enzymes and cytokeratins. Pancreatoblastoma lacks a prominent pseudopapillary pattern and expression of vimentin.
With a five-year survival rate of 97% after complete resection, often involving a radical procedure (PPPD or distal/partial pancreatectomy), the prognosis of SPTs is usually excellent [12]. Metastasis develops in up to 15% of patients, after an average disease-free interval of 8.5 years [3, 13]. Metastasis occurs mainly in the liver (often as a solitary deposit which may be amenable to surgical resection) and peritoneum, and rarely affects the regional lymph nodes. Even in the presence of metastatic disease at initial diagnosis, long-term survival is often the rule [14]. At present, there are no established histological criteria to predict the biological behaviour of SPTs. While invasion of blood vessels, perineural clefts or adjacent organs, a high degree of cellular pleopmorphism and an elevated mitotic rate are suggested to be associated with metastasis and recurrence, absence of these features does not preclude malignant behaviour. Thus, all patients require long-term follow-up. The poorly differentiated tumour component with an aberrant immunoprofile in Case 1 may relate to the unusually aggressive behaviour of this tumour. A recent study of two similarly aggressive cases also observed a tumour component with unusual histopathological features. There was a diffuse growth pattern, extensive tumour necrosis, significant nuclear atypia and an unusually high mitotic rate, and in one a component of sarcomatoid carcinoma. Immunoprofiles were similar to conventional SPTs except there was abnormal beta-catenin distribution and markedly increased MIB1 expression [15].
The benefit of chemotherapy or radiotherapy for patients with SPT is unknown. There is one documented case of radiosensitivity [16], and pre-operative chemotherapy has been reported as rendering the tumour resectable in at least two cases, using cis-platinum and 5- fluoruracil [17] or gemcitabine [18]. There is also a report of post-operative use of ifosfamid, cisplatin and VP16 in a metastasising SPT [19].
More than four decades after its initial description, the histogenesis of SPTs is still controversial. An acinar cell, endocrine, ductal, extrapancreatic and recently, based on expression of a melanocytic marker (HMB45), a neuroectodermal origin has been proposed [20]. In view of the diverse immunohistochemical expression pattern, derivation from a pancreatic stem cell has also been suggested. However, none of these hypotheses explain the strongly sex-linked occurrence of the SPTs. Recent molecular studies revealed that SPTs are genetically different from pancreatic ductal adenocarcinoma and almost always harbour beta-catenin mutations [21]. This finding raises the possibility that the reported association of SPTs with familial adenomatous polyposis (FAP) and FAP manifestations such as colon cancer and papillary thyroid carcinoma may not be merely coincidental [13, 22]. Deregulation of cell cycle proteins has recently been demonstrated, however, further studies are needed to correlate molecular aberrations with the biological behaviour of SPTs [23].
CONCLUSION
SPT is a rare tumour predominantly occurring in young females and thought to be of low malignant potential. We have described five very diverse cases. Case 1 was particularly unusual given its rapid fatal outcome, atypical morphology and aberrant immunoprofile. Case 2 was unusual because of the patient’s advanced age at presentation. Cases 3 and 5 were exceptional given their very young age. In conclusion, it is clear that there is great variability in the presentation and clinical course of SPTs. Further research is required to elucidate their histogenesis and to predict their biological behaviour.
References
- Frantz VK. Tumors of the pancreas. In: Atlas of Tumor Pathology, 1st series. Washington, DC, USA: US Armed Forces Institute of Pathology, 1959:32-3.
- Solcia E, Capella C, Kloppel G. Tumors of the pancreas. In: Atlas of Tumor Pathology, 3rd series, Fascicle 20. Washington, DC, USA: US Armed Forces Institute of Pathology, 1997:120-44.
- Lam KY, Lo CY, Fan ST. Pancreatic solid-cysticpapillary tumor: clinicopathologic features in eight patients from Hong Kong and review of the literature. World J Surg 1999; 23:1045-50. [PMID 10512945]
- Yoon DY, Hines OJ, Bilchik AJ, Lewin K, Cortina G, Reber HA. Solid and papillary epithelial neoplasms of the pancreas: aggressive resection for cure. Am Surg 2001; 67:1195-9. [PMID 11768829]
- Buetow PC, Buck JL, Pantongrag-Brown L, Beck KG, Ros PR, Adair CF. Solid and papillary epithelial neoplasm of the pancreas: Imaging-pathologic correlation in 56 cases. Radiology 1996; 199:707-11. [PMID 8637992]
- Bennett GL, Hann LE. Pancreatic ultrasonography. Surg Clin North Am 2001; 81:259-81. [PMID 11392416]
- Megibow AJ, Lavelle MT, Rofsky NM. The pancreas revisited I: Diagnosis, chronic pancreatitis. Surg Clin North Am 2001; 81:307-20. [PMID 11392418]
- Orlando CA, Bowman RL, Loose JH. Multicentric papillary-cystic neoplasm of the pancreas. Arch Pathol Lab Med 1991; 115:958-60. [PMID 1718242]
- Kloppel G, Maurer R, Hofmann E, Luthold K, Oscarson J, Forsby N, et al. Solid-cystic (papillarycystic) tumours within and outside the pancreas in men: report of two patients. Virchows Arch A Pathol Anat Histopathol 1991; 418:179-83. [PMID 1705067]
- Kim YI, Kim ST, Lee GK, Choi BI. Papillary cystic tumor of the liver. A case report with ultrastructural observation. Cancer 1990; 65:2740-6. [PMID 2340471]
- Bardales RH, Centeno B, Mallery JS, Lai R, Pochapin M, Guiter G, Stanley MW. Endoscopic ultrasound-guided fine-needle aspiration cytology diagnosis of solid-pseudopapillary tumor of the pancreas. Am J Clin Pathol 2004; 121:654-62. [PMID 15151205]
- Madan AK, Weldon CB, Long WP, Johnson D, Raafat A. Solid and papillary epithelial neoplasm of the pancreas. J Surg Oncol 2004; 85:193-8. [PMID 14991875]
- Gonzalez-Campora R, Rios Martin JJ, Villar Rodriguez JJ, Otal Salaverri C, Hevia Vazquez A, Valladolid JM, et al. Papillary cystic neoplasm of the pancreas with liver metastasis coexisting with thyroid papillary carcinoma. Arch Pathol Lab Med 1995; 119:268-73. [PMID 7887782]
- Martin RC, Klimstra DS, Brennan MF, Conlon KC. Solid-pseudopapillary tumor of the pancreas: a surgical enigma? Ann Surg Oncol 2002; 9:35-40. [PMID 11833495]
- Tang LH, Aydin H, Brennan MF, Klimstra DS. Clinically aggressive solid pseudopapillary tumors of the pancreas: a report of two cases with components of undifferentiated carcinoma and a comparative clinicopathologic analysis of 34 conventional cases. Am J Surg Pathol 2005; 29:512-9. [PMID 15767807]
- Fried P, Cooper J, Balthazar E, Fazzini E, Newall J. A role for radiotherapy in the treatment of solid and papillary neoplasms of the pancreas. Cancer 1985; 56:2783-5. [PMID 4052952]
- Strauss JF, Hirsch VJ, Rubey CN, Pollock M. Resection of a solid and papillary epithelial neoplasm of the pancreas following treatment with cis-platinum and 5-fluorouracil: A case report. Med Pediatr Oncol 1993; 21:365-7. [PMID 8492753]
- Maffuz A, The Bustamante F, Silva JA, Torres- Vargas S. Preoperative gemcitabine for unresectable, solid pseudopapillary tumour of the pancreas. Lancet Oncology 2005; 6:185-6. [PMID 15737835]
- Rebhandl W, Felberbauer FX, Puig S, Paya K, Hochschorner S, Barlan M, Horcher E. Solidpseudopapillary tumor of the pancreas (Frantz tumor) in children: report of four cases and review of the literature. J Surg Oncol 2001; 76:289-96. [PMID 11320522]
- Chen C, Jing W, Gulati P, Vargas H, French SW. Melanocytic differentiation in a solid pseudopapillary tumor of the pancreas. J Gastroenterol 2004; 39:579- 83. [PMID 15235877]
- Tanaka Y, Kato K, Notohara K, Hojo H, Ijiri R, Miyake T, et al. Frequent beta-catenin mutation and cytoplasmic/nuclear accumulation in pancreatic solidpseudopapillary neoplasm. Cancer Res 2001; 61:8401- 4. [PMID 11731417]
- Ruo L, Coit DG, Brennan MF, Guillem JG Longterm follow-up of patients with familial adenomatous polyposis undergoing pancreaticoduodenal surgery. J Gastrointest Surg 2002; 6:671-5. [PMID 12399055]
- Muller-Hocker J, Zietz CH, Sendelhofert A. Deregulated expression of cell cycle-associated proteins in solid pseudopapillary tumor of the pancreas. Mod Pathol 2001; 14:47-53. [PMID 11235905]







