Juan Yu, Minzhou Zhang* and Jianyong Qi*
Intensive Care Laboratory, Animal Laboratory, Guangdong Province Hospital of Chinese Medicine, Guangzhou, Guangdong Provincial Academy of Chinese Medicine
*Corresponding Authors:
Jianyong Qi
Intensive Care Laboratory
Guangdong Province Hospital of Chinese Medicine
Guoyi West Road, Panyu District
Guangzhou, China
Tel: 86-2013751859805
E-mail: minzhouzhang8@163.com
Minzhou Zhang
Intensive Care Laboratory
Guangdong Province Hospital of Chinese Medicine
Guoyi West Road, Panyu District
Guangzhou, China
Tel: 86-2013751859805
E-mail: minzhouzhang8@163.com
Received: April 12, 2016; Accepted: April 16, 2016; Published: April 20, 2016
Citation: Yu J, Zhang M, Qi J. Role of CaMKII in CaMKII/[Na+]i/[Ca2+]i Feedback in Myocardial Ischemia and Reperfusion Injury:A Simulation Study. Interv Cardiol J 2015, 2:1.
Myocardial ischemia and reperfusion injury (MIRI) is a serious complication after percutaneous coronary intervention, which leads to heart failure, increased infarction size, and severe arrhythmias [1,2]. Various signaling pathways were found to be participated in the process of MIRI, such as phosphatidylinositol 3-kinase (PI3K)/AKT, extracellular signal-regulated kinase (ERK), endothelial NO synthase (eNOS), etc[3-5]. Recently, more and more studies demonstrate that Na+/Ca2+/Calmodulin-dependent protein kinase II (CaMII) feedback plays vital role in heart failure [6]. Over-activity of CaMKII enhanced the late Na+ current(INaL), caused intracellular Na+ ([Na+]i) increasing, changing the equilibrium of the Na+-Ca2+ exchanger (NCX) to impair forwardmode (Ca2+ extrusion), and aggravating reverse-mode (Ca2+ influx) exchange. Subsequently, this Ca2+ overload further activate CaMKII and cause a feedback loop of CaMKII/[Na+]i/ [Ca2+]i in heart failure. However, the role of CaMKII/[Na+]i/[Ca2+] i feedback in MIRI is still unclear. In the present study, we took the published MIRI action potential model [7,8], 50% blocking the voltage-dependent sodium current(INa) and CaMKII, to observe the effect of INa(-) and CaMKII(-) on MIRI.
To ensure the influence of INa and CaMKII on MIRI, we used Roberts-Christini MIRI AP model [7,8], which combined the dynamic changes of intracellular pH (pHi), extracellular pH(pHe), 13 ion currents and 6 pump exchangers. All simulations were run using a pacing rate of 1 Hz. Four groups were set as: Control (MIRI only), INa 50% block, CaMKII 50% block and both (INa 50% and CaMKII 50% block), respectively.
As we showed in Figures 1A-1C, during ischemia stage, intracellular and extracellular pH were reduced to 5.8 and 6.2 from pre-ischemia stage(pHe 7.4, pHi 7.2), respectively. The AP amplitude was decreased alternatively, which were consistent with the previous report [9]. In Figures 1D-1G, we can see that sodium overload existed in 10-minutes ischemia stage, compared with pre-ischemia, exacerbating in 10-minutes reperfusion stage. INa 50% block (blue line) slightly reduced intracellular sodium concertration ([Na]i), while activated CaMKII 50% block (green line) and both block (black line) reduced [Na]i more significantly. Calcium overload were found in control (red line) and INa 50% block(blue line), while it was reduced significantly in CaMKII 50% block and both block (Figure 1H), which reveals that CaMKII is essential to keep calcium overload, not INa. Consistently, we found NCX current were also reduced in CaMKII 50% block and both block (Figure 1I), which suggested that NCX played downstream role of CaMKII and calcium overload.
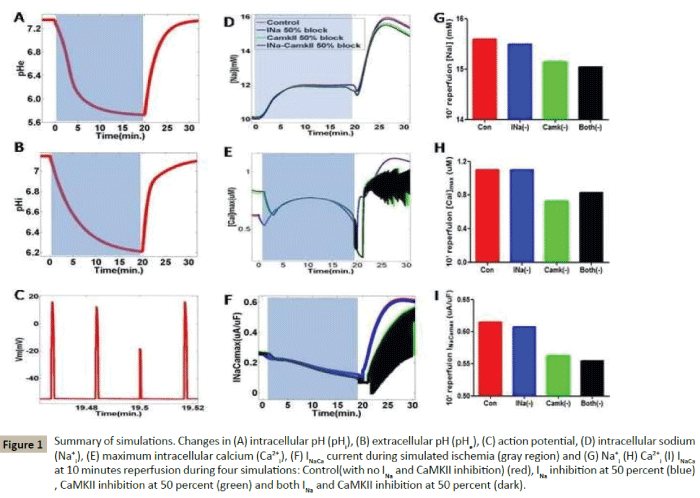
Figure 1: Summary of simulations. Changes in (A) intracellular pH (pHi), (B) extracellular pH (pHe), (C) action potential, (D) intracellular sodium (Na+i ), (E) maximum intracellular calcium (Ca2+i), (F) INaCa current during simulated ischemia (gray region) and (G) Na+i (H) Ca2+i (I) INaCaat 10 minutes reperfusion during four simulations: Control(with no INa and CaMKII inhibition) (red), INa inhibition at 50 percent (blue), CaMKII inhibition at 50 percent (green) and both INa and CaMKII inhibition at 50 percent (dark).
To further explore the mechanisms of CaMKII and INa on MIRI, we analysed various currents which might be related with sodium and calcium overload. As we showed in Figures 2A-2I, during preischemia stage, we can see that Peak INa keeps in 350 uA/uF in normal, while it reduced half (175 uA/uF) in INa 50% block and both groups. All other currents, such as calcium-sodium exchanger (ICaNa), INaL, background sodium current (INab), sodium-potassium exchanger (INaK), ATP-inactivated potassium current (IKatp), sodiumcalcium exchanger (INCX), sodium-bicarbonate symporter (INBC), sodium-exchanger (INHE), are nearly the same values in these four groups. During 10-minutes ischemia stage, Peak INa among the four groups were reduced significantly (40-60 uA/uF). Peak INab increased in CaMKII 50% block and both INa-CaMKII block groups, while all other currents kept the simular values among four groups. During 10-minutes of reperfusion stage, Peak ICaNa were significantly increased in CaMKII 50% block and both INa-CaMKII block groups, which reveals that more calcium ions were transferred from inside to outside of cells in these two groups, while other currents changed little. Therefore, ICaNa takes responsibility for reducing calcium overload in MIRI.
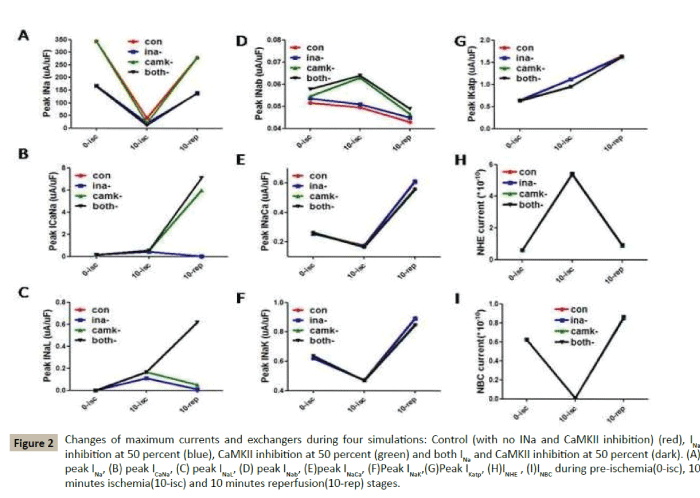
Figure 2: Changes of maximum currents and exchangers during four simulations: Control (with no INa and CaMKII inhibition) (red), INa inhibition at 50 percent (blue), CaMKII inhibition at 50 percent (green) and both INa and CaMKII inhibition at 50 percent (dark). (A) peak INa, (B) peak ICaNa, (C) peak INaL, (D) peak INab, (E)peak INaCa, (F)Peak INaK,(G)Peak IKatp, (H)INHE , (I)INBC during pre-ischemia(0-isc), 10 minutes ischemia(10-isc) and 10 minutes reperfusion(10-rep) stages.
As for the limitation, we used Roberts-Chritini MIRI AP model, which was the latest AP model describing the acidosis state and various currents' changes in MIRI, more information might be focused on the Markov state of ion channels.
Conclusion
The mechanisms of MIRI is complicated, considering the system is combined with various nonlinear components and frequently exhibit non-intuitive behavior, using the dynamic MIRI model showed in the present study will provide a powerful method to reveal the vague secrets. Here we used the MIRI model tof provide a solve to the interesting question, what is the focus of CaMKII/[Na+]i/[Ca2+]i feedback, which is more important between CaMKII and INa. Our study indicate that CaMKII plays a key role in calcium overload in MIRI, not INa, blocking of CaMKII activated INCX and ICaNa currents, transferred more Ca2+ to the outside of cell, subsequently reducing calcium overload, and finally alleviated MIRI. Inhibited activation of CaMKII might be a potential treatment in protection against myocardial ischemia and reperfusion injury.
Acknowledgments
This study was supported by grant No. 2014A030313402 from Guangdong Scientific Foundation, No. A2014271 from Guangdong Medical Research Foundation and Project of Wenxin herbs on AF from Guangdong science and Technology Foundation (to Jianyong Qi), Special Funds of Guangdong hospital of Chinese Medicine to Minzhou Zhang 2013,2014 and Jianyong Qi 2014. Special fund to Juan Yu from 2015 Guangdong Administration of Chinese Medicine
References
- Zalewski J, Claus P, Bogaert J, Driessche NV, Driesen RB, et al. (2015) Cyclosporine A reduces microvascular obstruction and preserves left ventricular function deterioration following myocardial ischemia and reperfusion.Basic Res Cardiol110: 18.
- Gonca E, Darıcı F (2015) The effect of cannabidiol on ischemia/reperfusion-induced ventricular arrhythmias: the role of adenosine A1 receptors.J Cardiovasc Pharmacol Ther 20: 76-83.
- Wang J, Yang H, Hu X, Fu W, Xie J, et al. (2013) Dobutamine-mediated heme oxygenase-1 induction via PI3K and p38 MAPK inhibits high mobility group box 1 protein release and attenuates rat myocardial ischemia/reperfusion injury in vivo. J Surg Res 183:509-516.
- Liu Z, Chen JM, Huang H, Kuznicki M, Zheng S, et al. (2016) The protective effect of trimetazidine on myocardial ischemia/reperfusion injury through activating AMPK and ERK signaling pathway.Metabolism 65:122-130.
- Guo Y, Li Q, Wu WJ, Tan W, Zhu X, et al. (2008) Endothelial nitric oxide synthase is not necessary for the early phase of ischemic preconditioning in the mouse. J Mol Cell Cardiol 44:496-501.
- Morotti S, Edwards AG, McCulloch AD, Bers DM, Grandi E (2014) A novel computational model of mouse myocyte electrophysiology to assess the synergy between Na+ loading and CaMKII. J Physiol592:1181-1197.
- Roberts BN, Christini DJ (2012) The relative influences of phosphometabolites and pH on action potential morphology during myocardial reperfusion: a simulation study.PLoS One7:e47117.
- Roberts BN, Christini DJ (2011) NHE inhibition does not improve Na(+) or Ca(2+) overload during reperfusion: using modeling to illuminate the mechanisms underlying a therapeutic failure.PLoS Comput Biol 7:e1002241.
- An J, Varadarajan SG, Camara A, Chen Q, Novalija E, et al. (2001) Blocking na(+)/h(+) exchange reduces [na(+)](i) and [ca(2+)](i) load after ischemia and improves function in intact hearts. Am J Physiol Heart CircPhysiol281: 2398–2409.