Shashikant Tiwari*
Department of Pediatrics, University of California, San Diego, CA, USA
*Corresponding Author:
Shashikant Tiwari
Department of Pediatrics, University of CaliforniaEpigenetic
San Diego, CA, USA.
Tel: 8584057372
E-mail: sktiwari@ucsd.edu
Received date: June 07, 2017; Accepted date: July 05, 2017; Published date: July 10, 2017
Citation: Tiwari S (2017) Recent Advancement in Methodology for Understanding Epigenetic Modifications. J Clin Epigenet. 3:21. doi: 10.21767/2472-1158.100055
Copyright: © 2017 Tiwari S. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Keywords
Epigenetics; DNA methylation; RNA methylation; Chromatin; Microarrays; 5-methylcytosine
Introduction
Epigenetics is the biological phenomenon in which biomolecules such as DNA, RNA and proteins are modified chemically or structurally without altering their primary sequence [1,2]. Epigenetics leads to stable heritable traits (phenotypes). Therefore, the epigenetic acts as a connection between the genotype and the phenotype. Various cellular processes such as gene expression, growth, cell cycle, development, disease progression, DNA replication and recombination are regulated by the epigenetic modifications [1]. Important epigenetic regulatory mechanisms include DNA methylation and hydroxy-methylation, histone modification, RNA methylation, and regulation by small and long noncoding RNAs [2]. These modifications can also dynamically change in response to specific diet, stress as well as cellular and environmental stimuli. When epigenetic mechanisms are misregulated, the result can be harmful to health and can lead to neurological disorders, cancer, cardiovascular disease and developmental irregularities [2,3]. Till date knowledge about how epigenetic changes take place and how they may contribute to development of specific diseases is at its infancy stage. This is due to insufficient availability of tools and technologies to characterize the epigenetic features of a cell on a global/ genome wide scale under normal conditions and during aging, development, and diseases. Thus, technologies would allow researchers to identify new molecular signatures and biomarkers of disease initiation, progression, and response to therapeutics. In this chapter, we highlight the methods utilized for understanding the complex mechanism of epigenetic regulation at genome wide levels. Further also address the role of computational and bioinformatics for answering the questions like how the epigenetic level senses the environmental stimuli during development and lineage specification and interaction among various chromatin modifications to control gene expression. This study uncovering the similarities and differences at epigenome level between individuals and their consequences in cellular gene regulation, differentiation and diseases. Therefore, epigenetic modifications are emerging as significant diagnostic and prognostic biomarkers in numerous fields of medicine.
DNA Methylation and Hydroxy- Methylation
DNA methylation is the most studied epigenetic mechanism which involved covalent addition of a methyl group (−CH3) to the fifth carbon of the pyrimidine ring of cytosine nucleotides. DNA methylation was performed by family of methyltransferase enzyme. The proteins comprise in this family which involved in de novo DNA methylation are DNMT3A and DNMT3B [4,5], while DNA methylation maintenance during replication is performed by DNMT1 [6]. DNA methylation plays a vital role during development and precise functioning of cells. Cellular genome is regulated by means of DNA methylation. During the past decades, enormous advances on tools to measure the level of DNA methylation have occurred. Nowadays techniques are available to measure DNA methylation at a single-base resolution in a reasonable manner.
In addition, another methylation common in human beings is 5-hydroxymethylcytosine (5hmC), which involved hydroxylation of the 5mC by the TET1 protein [7]. It has been play important role in neuronal development [8]. Further, it has been reported that 5hmC present in DNA of bacteriophage for the means of defense mechanism from restriction enzymes of the infected hosts [9,10]. Several methods have been developed to interrogate DNA methylations which are sodium bisulfite based, restriction enzyme based, affinity based, or based on physical properties. Some details of these methods and bioinformatic tools for analysis are summarized in Tables 1 and 2.
| Epigenetic modifications |
Experimental Approaches/Techniques |
Applications |
| DNA Methylation/Hydroxy-methylation |
High-performance liquid chromatography (HPLC) and matrix-assisted laser desorption–ionization time of flight (MALDI–TOF) mass spectrometry, Methylated DNA immunoprecipitation (MeDIP), Methylation sensitive Restriction Enzyme Sequencing (MRE-Seq), Whole-genome bisulfite sequencing (WGBS), DNA Methylation Microarrays |
To identify DNA methylation across parts of the genome at variable levels of resolution down to base pair level. |
| Histone modifications |
Chromatin Immunoprecipitation coupled with high-throughput sequencing (ChIP-seq) |
Applicable for genome- wide characterization and identification of functional elements such as transcription factors, components of the transcription machinery, specific histone modifications in the human genome. |
| |
Top-Down Mass Spectrometry |
Profiling changes in histone post translational modifications. |
| RNA Modifications |
RiboMeth-seq |
It is a sequencing-based method for profiling and quantitation of ribose methylation 2'-O-Me in RNA. |
| |
Bisulfite sequencing |
Methylation status of specific cytosines in RNAs through high-throughput sequencing techniques to studies at nucleotide resolution on a transcriptome-wide scale. |
| |
Liquid chromatography tandem mass spectrometry (LC-MS/MS). |
Analysis and detection of cellular Methylated RNAs. |
| |
Individual nucleotide resolution crosslinking immunoprecipitation (iCLIP) |
Immunoprecipitation-based transcriptomic methods for mapping of methyl-5- cytosine (m5C) or methyl- 6-adenosine (m6A) modification at nucleotide resolution in the human transcriptome. |
Table 1: Tools for detection of epigenetic modifications.
| |
Epigenetic Database |
Epigenetic Tools for Statistical Data Analysis and Visualization |
| Chromatin |
4D Genome: This is database of chromatin interactions from 3C, 4C, 5C, ChIA-PET, Hi- C, Capture-C, and IM- PET.
3CDB: A manually curated chromosome conformation capture (3C) database. Histome: Database for thesite of human histone-modifications, and histone modifying enzymes.
cisRED: A motif database. |
MACS: Model-based Analysis of ChIP-seq (MACS) is a peak-finding algorithm.
PAVIS: PAVIS (Peak Annotation and Visualization) helps to annotate and visualize ChIP-seq and BS-seq data.
chroGPS: A Bioconductor (R) package aimed at integration, visualization, and functional analysis of epigenomics data.
MMDiff: A Bioconductor (R) package that detect differential peaks in ChIP-seq data.
ChIP-Array v2.0: Integrate your ChIP-seq or ChIP- CHIP data with gene expression to build a regulatory network.
Epigenomix: A Bioconductor (R) package that integrate RNA-seq or microarray data with ChIP-seq data. Helps to preprocess and create differential gene lists for both data sets. |
| DNA Methylation |
MethylomeDB: Database provides DNA methylation profiles from humans and mice brain.
MethBase: Database for methylomes from well-studied organisms
NGSmethDB: Whole- genome bisulfite sequencing (WGBS) database.
DiseaseMeth: Methylomes of human disease. |
Bsseq: It is a Bioconductor (R) package for analyzing and visualizing WGBS data for identifying differentially methylated regions (DMRs).
MethPipe: For analysis of RRBS, and WGBS data to identify DMRs, allele-specific methylation, and partially methylated domains.
methylPipe: A Bioconductor (R) package for the analysis of CpG and non-CpG methylation from WGBS data.
epiGbs: It is a reference genome free reduced representation bisulfite sequencing (RRBS) method which allows cost-effective analysis of genetic variation and DNA methylation in large set of samples.
BEAT: A Bioconductor (R) package that lets you analyze single-cell BS-seq data. |
Table 2: Bioinformatic tools for analysis of epigenetic modifications.
Chromatin Remodeling
Chromatin remodeling is the dynamic modification of chromatin architecture to allow access of condensed genomic DNA to the regulatory transcription factors, through which they control expression of genes [11,12]. Chromatin remodeling also play important epigenetic regulatory role in various key biological processes such as apoptosis, DNA replication and repair, and in pluripotency [11]. Alteration in chromatin remodeling associated with various developmental disorders and cancers [12-14]. Currently targeting chromatin remodeling pathways is a major therapeutic strategy in the treatment of various diseases. There are mainly two ways through which chromatin remodeling takes place:
1. Histone modification.
2. ATP dependent modeling to restructure nucleosome. Histone modification is a covalent post-transcriptional modification of histone proteins by specific enzymes likewise methyltransferases, histone acetyltransferases (HATs), deacetylases and kinases. Histone proteins involved in packaging of DNA by wrapping around eight histone proteins. The type of the modification imparts by these enzymes in histone proteins include acetylation, methylation, ubiquitylation, phosphorylation, and sumoylation [15,16]. Histone modifications involved in regulation of diverse biological processes such as transcriptional activation/inactivation, chromosome packaging, cell cycle progression, apoptosis, differentiation, DNA replication, nuclear import, DNA damage/repair and neuronal repression [14]. Thus, quantitative detection of numerous histone modifications is important for understanding the epigenetic regulation of these various cellular processes and modeling the drug targets against histone modifying enzymes. Recent commonly used techniques for detection of histone modification and bioinformatics data base are summarized in Tables 1 and 2.
RNA Methylation
RNA Methylation occurs among all three kingdoms of life such as archaea, bacteria and eukaryotes and playing numerous biological functions such as intracellular trafficking, stress/ immune response, gene regulation, and RNA stabilization [17]. RNA Methylation typically takes place during post-transcriptional modifications and only few known to occur co-transcriptionally. It occurs in different RNAs including mRNA, rRNA, tRNA, snRNA, miRNA, snoRNA, and viral RNA. Diverse catalytic strategies are employed for RNA methylation by a variety of RNAmethyltransferases. Target of methylation sights mainly include the majority of nitrogens, except the nitrogens engaged in the glycosidic bond, and N7 and N3 position of adenosine [17]. Other site of modification includes the oxygen of the 2’OH moiety, the carbon atom at position 2 and in adenosine, the carbon atoms at position 5 in pyrimidines [17]. The most abundant and common RNA modifications in some viruses and most eukaryotes is N6-methyladenosine (m6A) which accounts for more than 80% of total RNA methylation [18-23]. In addition, it is also found in rRNA, tRNA, and small nuclear RNA (snRNA) as well as various long non-coding RNA, such as Xist [24,25]. It is well known that m6A broadly regulates embryonic development and cell fates. Recently discovered proteins such as 'writers', 'erasers' and 'readers' of this RNA chemical mark strongly indicate dynamic regulatory role like well-known reversible epigenetic modifications of DNA and histone proteins. This add new angle in post-transcriptional regulation of gene expression [26]. Adenosine methylation is induced by a complex of m6A methyltransferase containing subunits METTL3, METTL14, and Wilms tumor 1 associated protein (WTAP PMID 9409616. Dynamic and reversible nature of m6A in mRNA is due to m6A demethylase such as fat mass and obesity-associated protein (FTO) [27,28] and alkB homolog 5 (ALKBH5) [29]. The biological functions of m6A are mediated by a group of RNA binding proteins known as m6A readers which recognize the methylated adenine on RNA. The YT521-B homology (YTH) domain family of proteins (YTHDF1, YTHDF2, YTHDF3 and YTHDC1) are direct m6A readers and have a conserved m6A-binding pocket [25,30-32]. The schematic representation of m6A methyltransferase complex shown in Figure 1. These m6A methyltransferases (writers) with m6A readers, together with demethylases (erasers), create a complex mechanism of m6A regulation in which erasers and writers determine the distributions of m6A on RNA, whereas readers mediate m6A-dependent functions. Another RNA modification is 5-methylcytosine (5-mC) which is also an epigenetic mark. The details of the methods utilized for the understanding the different types RNA modifications are summarized in Table 1 [33-42].
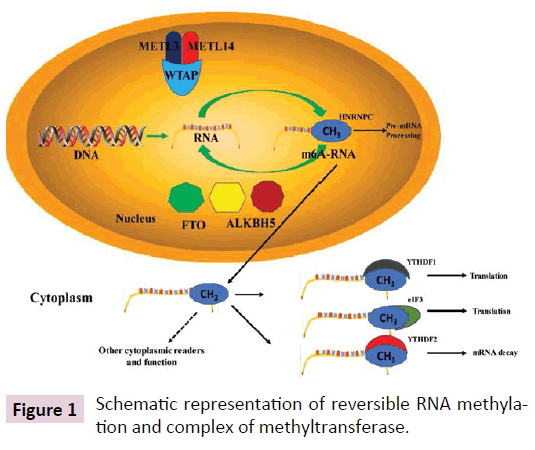
Figure 1: Schematic representation of reversible RNA methylation and complex of methyltransferase.
Single Cell Epigenomics
Most of epigenetic study involved the comprehensive epigenetic map of diverse array of cells and tissues. This result in providing the complete catalogue of average profile of the diverse cell types [43,44]. Average profile of diverse cell types fails to describe the study of rare cell types, poorly characterized cell populations and tissues which very hard to separate mixture of cell types [43,44]. Thus single cell epigenomics overcome these limitations and open a unique direction for epigenetic research. The commonly used recent emerging single-cell epigenomic techniques are described in Table 3 [45-49].
| Techniques |
Epigenetic modifications |
Methods |
Single Approach cell |
| DNA Methylation |
5mC |
BS-seq, MeDIP-seq |
Possible Currently No |
| 5hmC |
TAB-seq, hMeDIP-seq |
Possible Currently No |
| DNA-protein interactions |
Histone modifications |
ChIP-seq |
Possible |
| Chromatin structure |
Positioning of nucleosome
DNA accessibility |
MNase-seq and NOME-seq ATAC-seq |
Possible |
Table 3: Commonly used recent emerging single-cell epigenomic techniques.
Conclusion
Epigenetic modifications play a critical role in cell development and homeostasis. Its interrogation in many clinical situations has permitted us to the identification of various fundamental alterations of different diseases, such as cancer, neurodegenerative as well as neurodevelopmental disorders. On the other hand, it has also permit the detection of biomarkers for predicting disease progression and for diagnosis and response to treatments of diseases. Further heterogenous tissues and complex cell types can be studied by single cell epigenomic to understand the cellular plasticity and diversity, as seen in various stem cells and in diseases like cancer. Such advancement has been only possible by availability of highly developed tools and techniques for determining the various types of epigenetic modifications.
References
- Reik W (2007) Stability and flexibility of epigenetic gene regulation in mammalian development. Nature 447: 425-432.
- Berger SL, Kouzarides T, Shiekhattar R, Shilatifard A (2009) An operational definition of epigenetics. Genes Dev 23: 781-783.
- Lappalainen T, Greally JM (2017) Associating cellular epigenetic models with human phenotypes. Nat Rev Genet 18: 441-451.
- Okano M, S Xie, Li E (1998) Cloning and characterization of a family of novel mammalian DNA (cytosine-5) methyltransferases. Nat Genet 19: 219-220.
- Okano M, Bell DW, Haber DA, Li E (1999) DNA methyltransferases Dnmt3a and Dnmt3b are essential for denovo methylation and mammalian development. Cell 99: 247-57.
- Baylin SB, Jones (2011) A decade of exploring the cancer epigenome-biological and translational implications. Nat Rev Cancer 11: 726-734.
- Lorsbach RB, Moore J (2003) TET1, a member of a novel protein family, is fused to MLL in acute myeloid leukemia containing the t(10;11)(q22;q23). Leukemia 17: 637-641.
- Guibert S, Weber M (2013) Functions of DNA methylation and hydroxymethylation in mammalian development. Curr Top Dev Biol 104: 47-83.
- Wyatt GR, Cohen SS (1952) A new pyrimidine base from bacteriophage nucleic acids. Nature 170: 1072-1073.
- Vrielink A, Rüger W, Driessen HP, Freemont PS (1994) Crystal structure of the DNA modifying enzyme beta-glucosyltransferase in the presence and absence of the substrate uridine diphosphoglucose. Embo J 13: 3413-3422.
- Lorch Y, Maier-Davis B, Kornberg RD (2010) Mechanism of chromatin remodeling. Proc Natl Acad Sci U S A 107: 3458-3462.
- Boeger H, Griesenbeck J, Kornberg RD (2008) Nucleosome retention and the stochastic nature of promoter chromatin remodeling for transcription. Cell 133: 716-726.
- Lorch Y, Maier-Davis B, Kornberg R (2014) Role of DNA sequence in chromatin remodeling and the formation of nucleosome-free regions. Genes Dev 28: 2492-2497.
- Jenuwein T, Allis CD (2001) Translating the histone code. Science 293: 1074-1080.
- Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, et al. (2007) High-resolution profiling of histone methylations in the human genome. Cell 129: 823-837.
- Strahl BD, Allis CD (2000) The language of covalent histone modifications. Nature 403: 41-45.
- Motorin Y, Helm M (2011) RNA nucleotide methylation. Wiley Interdiscip Rev RNA 2: 611-631.
- Beemon K, Keith J (1977) Localization of N6-methyladenosine in the Rous sarcoma virus genome. J Mol Biol 113: 165-179.
- Aloni Y, Dhar R, Khoury G (1979) Methylation of nuclear simian virus 40 RNAs. J Virol 32: 52-60.
- Desrosiers R, Friderici K, Rottman F (1974) Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. Proc Natl Acad Sci U S A 71: 3971-3975.
- Perry RP, Kelley DE (1975) The methylated constituents of L cell messenger RNA: evidence for an unusual cluster at the 5' terminus. Cell 4: 387-394.
- Zhong S, Li H (2008) MTA is an Arabidopsis messenger RNA adenosine methylase and interacts with a homolog of a sex-specific splicing factor. Plant Cell 20: 1278-1288.
- Bodi Z, Button JD, Grierson D, Fray RG (2010) Yeast targets for mRNA methylation. Nucleic Acids Res 38: 5327-5335.
- Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, et al. (2012) Comprehensive analysis of mRNA methylation reveals enrichment in 3' UTRs and near stop codons. Cell 149: 1635-1646.
- Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, et al. (2012) Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 485: 201-206.
- Fu Y, Dominissini D, Rechavi G, He C (2014) Gene expression regulation mediated through reversible m(6)A RNA methylation. Nat Rev Genet 15: 293-306.
- He C (2010) Grand challenge commentary: RNA epigenetics. Nat Chem Biol 6: 863-865.
- Jia G, Fu Y, Zhao X, Dai Q, Zheng G, et al. (2011) N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol 7: 885-887.
- Zhong S, Li H, Bodi Z, Button J, Vespa L, et al. (2013) ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell 49: 18-29.
- Wang X, Lu Z, Gomez A, Hon GC, Yue Y, et al. (2014) N6-methyladenosine-dependent regulation of messenger RNA stability. Nature 505: 117-120.
- Wang X, Zhao BS (2015) N(6)-methyladenosine modulates messenger rna translation efficiency. Cell 161: 1388-1399.
- Xiao W, Adhikari S, Dahal U, Chen YS, Hao YJ, et al. (2016) Nuclear m(6)A reader YTHDC1 regulates mRNA splicing. Mol Cell 61: 507-519.
- Harris RA, Wang T, Coarfa C, Nagarajan RP, Hong C, et al. (2010) Comparison of sequencing-based methods to profile DNA methylation and identification of monoallelic epigenetic modifications. Nat Biotechnol 28: 1097-105.
- Moran S (2017) Technologies for Deciphering Epigenomic DNA Patterns. Adv Exp Med Biol 978: 477-488.
- Marinov GK (2017) ChIP-seq for the identification of functional elements in the human genome. Methods Mol Biol 1543: 3-18.
- Zhu J, Adli M, Zou JY, Verstappen G, Coyne M, et al. (2013) Genome-wide chromatin state transitions associated with developmental and environmental cues. Cell 152: 642-654.
- Zhou M, Wu S, Stenoien DL, Zhang Z, Connolly L, et al. (2017) Profiling Changes in Histone Post-translational Modifications by Top-Down Mass Spectrometry. Methods Mol Biol 1507: 153-168.
- Krogh N, Birkedal U, Nielsen H (2017) RiboMeth-seq: profiling of 2'-O-Me in RNA. Methods Mol Biol 1562: 189-209.
- Amort T (2015) Transcriptome-wide detection of 5-Methylcytosine by Bisulfite sequencing. Methods Mol Biol 1562: 123-142.
- Thüring K, Schmid K, Keller P, Helm M (2017) LC-MS analysis of methylated RNA. Methods Mol Biol 1562: 3-18.
- George H, Ule J, Hussain S (2017) Illustrating the epitranscriptome at nucleotide resolution using methylation-iCLIP (miCLIP). Methods Mol Biol 1562: 91-106.
- Linder B, Grozhik AV, Olarerin-George AO, Meydan C, Jaffrey SR, et al. (2015) Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat Methods 12: 767-772.
- Clark SJ, Lee HJ, Smallwood SA, Kelsey G, Reik W, et al. (2016) Single-cell epigenomics: powerful new methods for understanding gene regulation and cell identity. Genome Biol 17: 72.
- Schwartzman O, Tanay A (2015) Single-cell epigenomics: techniques and emerging applications. Nat Rev Genet 16: 716-726.
- Farlik M, Sheffield NC, Nuzzo A, Datlinger P, Schönegger A, et al. (2015) Single-cell DNA methylome sequencing and bioinformatic inference of epigenomic cell-state dynamics. Cell Rep 10: 1386-1397.
- Guo H, Zhu P, Wu X, Li X, Wen L, et al. (2013) Single-cell methylome landscapes of mouse embryonic stem cells and early embryos analyzed using reduced representation bisulfite sequencing. Genome Res 23: 2126-2135.
- Rotem A, Ram O, Shoresh N, Sperling RA, Goren A, et al. (2015) Single-cell ChIP-seq reveals cell subpopulations defined by chromatin state. Nat Biotechnol 33: 1165-1172.
- Cusanovich DA, Daza R, Adey A, Pliner HA, Christiansen L, et al. (2015) Multiplex single cell profiling of chromatin accessibility by combinatorial cellular indexing. Science 348: 910-914.
- Buenrostro JD, Wu B, Litzenburger UM, Ruff D, Gonzales ML, et al (2015) Single-cell chromatin accessibility reveals principles of regulatory variation. Nature 523: 486-490.

