Keywords
Cell Proliferation; Insulin- Secreting Cells; Prolactin; Signal Transduction
Abbreviations
GH: growth hormone; Glc: glucose; GRB2: growth factor bound protein 2; HGF: hepatocyte growth factor; MAPK: mitogen activated protein kinase; MEK: MAPK kinase; mSOS: murine sons of sevenless-1 protein; IGF-I: insulin like growth factor-I; IRS: insulin receptor substrate; JAK: janus kinase; PI3’K: phosphatidylinositol-3’- kinase; PKA: protein kinase A; PKB: protein kinase B = AKT; PKC: protein kinase C; PKG: protein kinase G; Prl: prolactin; PY: anti-phosphotyrosine; p70S6k: 70kD S6 kinase; Shc: SH2-containing protein; STAT: signal transducer and activator of transcription.
INTRODUCTION
Pancreatic beta-cells are terminally differentiated and display a low mitotic index [1, 2]. Nevertheless, beta-cell proliferation is affected by a plethora of nutrients, hormones and growth factors. Due to this abundance of regulatory inputs, control of the beta-cell proliferation at the molecular level is highly complex. In particular, the response to certain hormonal ligands is highly dependent on the nutritional state of the cell [3, 4].
Under certain conditions, such as pregnancy and obesity, a low pancreatic beta-cell proliferation rate can increase considerably. Certain nutrients such as aminoacids or glucose, as well as changes in electrolyte concentrations (e.g. calcium ions), are known to increase the mitotic index of primary pancreatic beta-cells from 0.5 to 6%, and to even higher ratios in pancreatic beta-cell lines [2, 5, 6, 7,]. By contrast, an inhibitory effect on beta-cell proliferation has been observed in the presence of free fatty acids [8]. Furthermore, growth factors such as insulinlike growth factor (IGF-I), growth hormone (GH) or hepatocyte growth factor (HGF) have been shown to considerably enhance the numbers of replicating beta-cells in rodent islets [2, 9, 10].
The intracellular events modulating mitogenic signal transduction in pancreatic beta-cells in the presence of growth factors still remain largely unknown. IGF-I and GH stimulate beta-cell proliferation in a glucose-dependent fashion [6, 7] whereas HGF also stimulates cell proliferation glucose independently [11].
IGF-I stimulates beta-cell proliferation mainly via tyrosine autophosphorylation activation of the IGF-I-receptor tyrosine kinase which, in turn, results in a downstream tyrosine phosphorylation of insulin receptor substrate- 2 (IRS-2) and a subsequent activation of phosphatidylinositol-3’-kinase (PI3’K), as well as protein kinase B (PKB), mammalian target of rapamycin (mTOR) and 70 kD S6 kinase (p70S6k) [7, 12]. In contrast, the effect of GH and HGF is initiated via the activation of janus kinase 2 (JAK2) and the consecutive activation of the signal transducer and activator of transcription 5 (STAT 5) [6, 11]. Since growth factors initiate a plethora of signaling cascades, combinations of growth factors may act synergistically, as we have demonstrated previously, e.g. for IGF-I and GH [6].
Aside from being a required nutrient for pancreatic beta-cell growth, glucose initiates many signal transduction pathways. Glucose concentrations also modulate the proliferative response to other hormonal ligands [6, 11]. Therefore, glucose induces the activation of IRS-4 [11] and protein kinase C [7, 11, 13], and triggers the elevation of intracellular Ca2+ and cAMP concentrations [2, 5, 14]. In contrast, the involvement of mitogen activated protein kinase (MAPK) is only observed at low, subphysiological concentrations of glucose [6, 7, 12].
Prolactin is a hormone which is a strong candidate involved in the increase of pancreatic beta-cell proliferation observed during pregnancy. Production and secretion of prolactin is elevated in pregnancy, and the hormone has been recognized as a potent growth factor for pancreatic beta-cells [9, 15, 16]. Furthermore, it has been demonstrated that prolactin activates the JAK/STAT signaling pathway in pancreatic beta-cells [16, 17, 18]. However, our knowledge of other signaling pathways involved in prolactin-stimulated pancreatic beta-cell proliferation is still incomplete.
INS-1 cells secrete insulin in a glucose dependent manner [19]. The proliferation of this cell line is stimulated by nutrients and several growth factors [6, 7]. Thus, the INS-1 pancreatic beta-cell line is a well-defined model for the study of cell proliferation in insulin-producing cells. We herein report on the molecular signaling events which are triggered by the mitogenic action of prolactin in INS-1 cells.
METHODS
Materials
Methyl[3H]thymidine (20 Ci/mmol) was purchased from Dupont/NEN (Boston, MA, USA). Prolactin, IGF-I, GH and all protein activity inhibitors came from Calbiochem- Novabiochem (La Jolla, CA, USA). Antiphospho- MAPK antiserum was purchased from Promega Corp. (Madison, WI, USA); all the other antisera were from Upstate Biotechnology (Lake Placid, NY, USA). Transblot nitrocellulose membrane (0.45 μm pore size) was from Biorad (Hercules, CA, USA), Chemiluminescence detection kit (ECL+) was from Amersham (Buckingham, England). All the other chemicals were purchased from Sigma (Providence, RI, USA) or Merck AG (Darmstadt, Germany) and were of the highest purity available.
Cell Culture
The glucose sensitive pancreatic beta-cell line INS-1 was used for all experiments [19]. INS- 1 cells were maintained at 37°C, 5% CO2 in RPMI 1640 medium containing 50 μM beta mercaptoethanol, 1 mM sodium pyruvate, 2 mM L-glutamine, 10% fetal calf serum, 11.2 mM glucose, 100 units/mL penicillin, 100 μ g/mL streptomycin as described [19] and were subcultured at 80% confluence.
[3H]Thymidine Incorporation
INS-1 cell proliferation was quantified using [3H]thymidine incorporation [14, 20]. [3H]thymidine incorporation was more suited for the assessment of INS-1 cell proliferation than other methods (e.g., cell counting because INS-1 cells are adherent and are not easy to separate). The INS-1 cells (105/well) were subcultured in 96-well plates and incubated for 48 h to allow the cells to attach. The cells were made quiescent by starving them for 24 hours, replacing the medium with glucose free RPMI 1640 containing 0.1% BSA instead of serum. Cell growth was then stimulated for 24 hours by different glucose concentrations (0-24 mM) with or without prolactin (0.5-2 nM), plus/minus insulin-like growth factor I (IGF-I, 10 nM), plus/minus growth hormone (GH, 5-20 nM) with or without various protein activity inhibitors. Five μCi/mL [3H]thymidine was added for the last 4 hours of the incubation period to assess the proliferation rate of the INS-1 cells. The cells were then lysed using a semiautomatic cell harvester (Inotech, Dietikon, Switzerland) and the lysates were transferred to glass fiber micropore filters (Packard, Meriden, CT, USA). INS-1 cell DNA was trapped on the filters and the incorporated [3H]thymidine was counted by liquid scintillation counting method. All experiments were done in triplicate.
Protein Preparation
INS-1 cells were subcultured in 10cm dishes to about 60% confluence and made quiescent by glucose and serum deprivation for 24 h as described above. The cells were then incubated in fresh RPMI 1640 medium containing 0-24 mM glucose with our without 0.5-2 nM prolactin for 5-60 min and the cells then lysed in 0.5 mL of ice-cold lysis buffer (50mM Hepes, pH 7.5, 1% (v/v) nonidet P40, 2 mM sodium vanadate, 50 mM sodium fluoride, 10 mM sodium pyrophosphate, 4 mM EDTA, 10 μM leupeptin, 10 μg/mL aprotinin, and 100 μM phenylmethylsulfonyl fluoride) as described previously [21, 22]. Cell lysates were stored at -80°C. Protein concentrations were determined using the bicinchoninic acid method (Pierce, Rockford, IL, USA).
Protein Immunoblot and Co- Immunoprecipitation Analysis
Mitogenic signal transduction protein expression and protein tyrosine phosphorylation were tested using immunoblot analysis. Stimulated proteinprotein interactions between mitogenic signal transduction proteins were tested using coimmunoprecipitation analysis as described previously [21, 22]. Horseradish peroxidase based chemiluminescence reaction was used as the secondary detection method. Fifty to 75 μg of INS-1 cell total protein lysate were used for immunoblot analysis and 750 μg for coimmunoprecipitation analysis. Experiments were done at least three times. Quantification was performed by densitometric scanning with Bio-1D® (Vilber Lourmat, Eberhardzell, Garmany) software analysis and is presented as the sum of intensities (109 pixel).
STATISTICS
Data are presented as mean±SE of at least 5 independent experiments. Statistically significant differences between groups were analyzed by using the Student’s t test. Twotailed P values of less than 0.05 were considered statistically significant. Statistical analysis was carried out by using MS Excel software.
RESULTS
Prolactin Stimulates the Proliferation of INS-1 Cells in the Presence of Physiological Glucose Concentrations
[3H]thymidine incorporation was used to determine INS-1 cell proliferation after stimulation with different glucose concentrations (0-15 mM) with or without 0.5-2.0 nM prolactin. INS-1 cells were used as a model to examine pancreatic beta-cell proliferation, as they respond, unlike the majority of other pancreatic beta-cell lines, to glucose in the physiological relevant range (6- 18 mM glucose) in terms of insulin secretion [19] and cell proliferation [6, 7] with no indication of dedifferentiation [6, 7, 11].
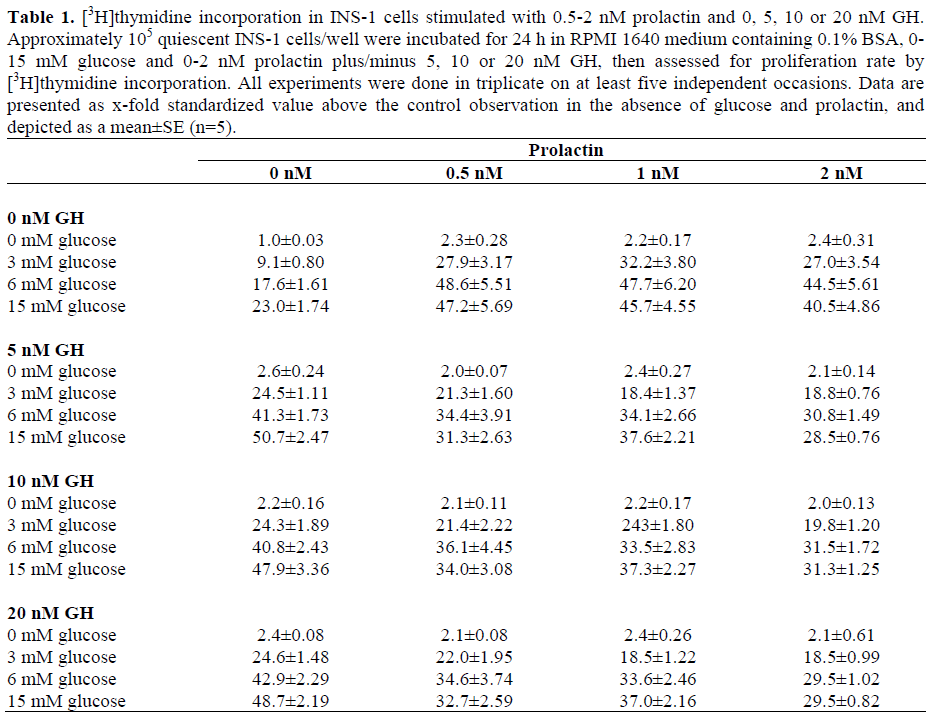
Prolactin (0.5-2 nM) stimulated INS-1cell proliferation up to 2.4-fold even in the absence of glucose (Figure 1). In the presence of physiological glucose concentrations (6 mM), the same effect is seen (Figure 1). Glucose in concentrations from 3 to 15 mM stimulated INS-1 cell proliferation in a concentration-dependent manner up to 23- fold compared to unstimulated controls, with a maximum at 15 mM glucose (Figure 2b). Co-stimulation with glucose and 0.5-2 nM prolactin further increased cell proliferation. Maximum stimulation (a 48.6-fold increase) was observed in the presence of 6 mM glucose and 0.5 nM prolactin, whereas no significant additional effect could be demonstrated upon higher doses of prolactin; instead, a trend towards reduced proliferation was observed. The effect was not significant (P=0.960 for 2 nM prolactin) and, at even higher prolactin concentrations, the proliferative effect lessened (data not shown). The additional stimulatory effect of prolactin at 0.5 surpassed the effect of glucose alone up to 3.5-fold (Figure 2a, P<0.001) with a maximum in the presence of 3 mM glucose. Insulin secretion/total protein in prolactin stimulated cells was slightly but not significantly increased (1.358 ng/mg total protein versus 1.128 ng/mg in non prolactin controls). Thus, the observed proliferative effect was not due to increased insulin secretion.
Figure 1. Different prolactin concentrations stimulate
[3H]thymidine incorporation in INS-1 cells at different
glucose concentrations. Approximately 105 quiescent
INS-1 cells/well were incubated for 24 h in RPMI 1640
medium containing 0.1% BSA, 0 or 6 mM glucose
plus/minus 0.5-2 nM prolactin, then assessed for
proliferation rate by [3H]thymidine incorporation. All
experiments were done in triplicate on at least eight
independent occasions. The data are presented as xfold
standardized value above the control observation
in the absence of prolactin and glucose (i.e. 3,000-
4,000 cpm/105 cells), and depicted as a mean±SE
(n=8).
* Significant differences (P<0.05) vs. control
observation in the absence of prolactin.
Figure 2. Prolactin-dependent [3H]thymidine
incorporation in INS-1 cells at different glucose
concentrations. Approximately 105 quiescent INS-1
cells/well were incubated for 24 h in RPMI 1640
medium containing 0.1% BSA, 0-15 mM glucose
plus/minus 0.5 nM prolactin, then assessed for
proliferation rate by [3H]thymidine incorporation. All
experiments were done in triplicate on at least eight
independent occasions. The data are presented as xfold
standardized value above the control observation
in the absence of glucose and prolactin (i.e. 3,000-
4,000 cpm/105 cells), and depicted as a mean±SE
(n=8). Referred significant changes are marked (*). a. Standardized value in [3H]thymidine incorporation in
prolactin-stimulated INS-1 cells at different glucose
concentrations versus only glucose-stimulated cells. b. Standardized value versus unstimulated controls.
* Significant differences (P<0.05)
As compared to combined stimulation with 0.5 nM prolactin and 6 mM glucose (48.6- fold increase, Figure 2), additional stimulation with 10 nM IGF-I did not result in an additional increase of [3H]thymidine incorporation (44-fold increase with glucose plus prolactin plus IGF-I relative to completely unstimulated controls), but rather yielded a slightly diminished proliferation, even though this reduction was not significant (P=0.313) (Figure 3). Co-stimulation of INS-1 cells with 6 mM glucose, 0.5-2 nM prolactin, and 5-20 nM rat growth hormone (GH) lead to an almost significant (P=0.053) GHdependent reduction in INS-1 cell proliferation at all GH-concentrations as compared to stimulation with glucose and prolactin alone (Figure 3, Table 1). The reduction was also dependent on the glucose concentration. [3H]thymidine incorporation in the presence of 20 nM GH in addition to 15 mM glucose and 2 nM prolactin was reduced to merely about 60% of the maximal stimulation observed at 6 mM glucose and 0.5 nM prolactin (29-fold increase compared to unstimulated controls; Table 1).
Figure 3. [3H]thymidine incorporation in INS-1 cells
stimulated with 0.5 or 2nM prolactin with or without
additional growth factors. Approximately 105 quiescent
INS-1 cells/well were incubated for 24 h in RPMI 1640
medium containing 0.1% BSA, 6 mM glucose
plus/minus 0.5 or 2 nM prolactin plus/minus 10 nM
IGF-1 plus/minus 10 nM GH, then assessed for
proliferation rate by [3H]thymidine incorporation. All
experiments were done in triplicate on at least five
independent occasions. Data are presented as x-fold
standardized value above the control observation in the
absence of glucose and prolactin, and depicted as a
mean±SE (n=5).
* Significant differences (P<0.05)
Prolactin Dependent Mitogenic Signal Transduction Pathways in INS-1 Cells
The protein phosphorylation-dependent activation of mitogenic signal transduction pathways by 1 nM prolactin plus/minus 3, 6 and 15 mM glucose in INS-1 cells was investigated using co-immunoprecipitation and immunoblot analysis. Immunoprecipitation with an anti-phosphotyrosine (PY) antibody followed by immunoblot analysis with IRS-1 antiserum showed an increase in phosphotyrosine phosphorylation of IRS-1 after a 10 min exposure to 1 nM prolactin in the presence of high glucose concentrations (15 mM) as compared to stimulation with 15 mM glucose alone or co-stimulation with 1 nM prolactin and low glucose concentrations (3 mM) (Figure 4). Immunoblot analysis for IRS-2 showed tyrosine phosphorylation even in the absence of glucose (especially if prolactin is added) with maximum activation in the presence of prolactin (1 nM) plus low glucose (3 mM) (Figure 5). IRS-2 phosphorylation lessened in the presence of higher glucose levels. IRS-4 yielded the strongest activation at 1 nM prolactin plus 6mM glucose, but also at low glucose levels. Thus, prolactin seems to activate the IRS proteins in the presence of different glucose concentrations.
Figure 4. Prolactin increases IRS-1 activation mainly
in the presence of high glucose concentrations. IRS-1
activation was examined in INS-1 cells (80% confluent
on a 15-cm diameter dish) after a treatment with 0- 3, 6
or 15 mM glucose plus/minus 1 nM prolactin for 10
min. Cell lysates were subjected to immunoprecipitation
(i.p.) with antiserum against phosphotyrosine
residues (PY) (a.) or against the p85
regulatory subunit of PI3‘K (b.). Immunoprecipitates
were then subjected to immunoblot (i.b.) analysis with
anti-IRS-1 antibodies. Experiments were done at least
three times. A representative blot for coimmunoprecipitation
analysis is shown. Quantification
was performed by densitometric scanning with Bio-
1D® (Vilber Lourmat, Eberhardzell, Garmany)
software analysis and is presented as sum of intensities
(109 pixel).
Figure 5. Prolactin increases IRS-2 activation mainly
in the presence of low glucose concentrations. INS-1
cells (80% confluent on a 15-cm diameter dish) were
stimulated with 0. 3, 6 or 15 mM glucose plus/minus 1
nM prolactin for 10 min. Cell lysates were subjected to
immunoprecipitation (i.p.) with antiserum against
phosphotyrosine residues (PY) (a.) or against the p85
regulatory subunit of PI3‘K (b.). Immunoprecipitates
were then subjected to immunoblot (i.b.) analysis with
anti-IRS-2 antibodies. Experiments were done at least
three times. A representative blot for coimmunoprecipitation
analysis is shown. Quantification
was performed by densitometric scanning with Bio-
1D® (Vilber Lourmat, Eberhardzell, Garmany)
software analysis and is presented as the sum of
intensities (109 pixel).
Immunoprecipitation of the 85 kDa regulatory subunit of PI3’K (PI3’K p85) followed by immunoblot analysis with IRS-1 antiserum also showed increased PI3’K binding in the presence of prolactin at all glucose concentrations with maximum binding in the presence of 1 nM prolactin and 15 mM glucose (Figure 4). In contrast, PI3’K-IRS-2 binding showed a maximum in the presence of prolactin plus low glucose concentrations (Figure 5) which emphasizes the effect seen in anti-phosphotyrosine phosphorylation. IRS-4 showed maximum association in the presence of physiological glucose concentrations (6 mM plus 1 nM prolactin; Figure 6) but also in the presence of low glucose concentrations (3 mM).
Figure 6. Prolactin increases the association of PI3‘K
with IRS-4 especially in the presence of physiological
glucose concentrations. INS-1 cells (80% confluent on
a 15-cm diameter dish) were stimulated with 0, 3, 6 or
15 mM glucose plus/minus 1 nM prolactin for 10 min.
Cell lysates were subjected to immunoprecipitation
(i.p.) with antiserum against the p85 regulatory subunit
of PI3‘K. Immunoprecipitates were then subjected to
immunoblot (i.b.) analysis with anti-IRS-4 antibodies.
Experiments were done at least three times. A
representative blot for co-immunoprecipitation analysis
is shown. Quantification was performed by
densitometric scanning with Bio-1D® (Vilber Lourmat,
Eberhardzell, Garmany) software analysis and is
presented as the sum of intensities (109 pixel).
Immunoprecipitation with anti-phosphotyrosine antibody, followed by immunoblot analysis with anti-JAK-2 antisera showed increased JAK-2 phosphorylation and activation after stimulation with 1 nM prolactin especially in the presence of physiological and subphysiological glucose concentrations (Figure 7) suggesting prolactin mediated activation of the JAK/STAT pathway in INS-1 cells as proposed before [15, 18, 23, 24]. Immunoprecipitation with PY prolactinand immunoblotting with STAT- 5 also showed increased phosphorylation activation in the presence of prolactin and physiological glucose concentrations (Figure 7).
Figure 7. Prolactin increases protein phosphotyrosine
phosphorylation of JAK-2 and STAT-5 glucose
dependently. INS-1 cells (80% confluent on a 15-cm
diameter dish) were stimulated with 0, 3, 6 or 15 mM
glucose plus/minus 1 nM prolactin for 10 min. Cell
lysates were subjected to immunoprecipitation (i.p.)
with antiserum against phosphotyrosine residues (PY).
Immunoprecipitates were then subjected to
immunoblot (i.b.) analysis with anti-JAK-2 (a.) or anti-
STAT-5 antibodies (b.). Experiments were done at
least three times. A representative blot for coimmunoprecipitation
analysis is shown. Quantification
was performed by densitometric scanning with Bio-
1D® (Vilber Lourmat, Eberhardzell, Garmany)
software analysis and is presented as the sum of
intensities (109 pixel).
Immunoblot analysis with specific antibodies against phosphorylated MAPK did not show significant phosphorylation after stimulation with prolactin (data not shown) whereas growth factor bound protein 2 (GRB2) showed an increased binding to SH2- containing protein (Shc) in the presence of prolactin at any glucose concentration (data not shown). These results indicate that the MAPK pathway may not play an important role in the prolactin-dependent proliferation of INS-1 cells, but prolactin seems to activate an alternate pathway which is activated by GRB2.
Effect of Protein Phosphorylation Inhibitors on Glucose and Prolactin- Stimulated INS-1-Cell Proliferation
The effect of various specific protein phosphatase and kinase inhibitors on [3H]thymidine incorporation was examined in the presence of 0. 3 and 15 mM glucose plus/minus 1 nM prolactin plus/minus 10 nM IGF-I (Table 2). Inhibition of tyrosine phosphatase by sodium orthovanadate (0.5 mM) completely inhibited glucose/prolactindependent cell proliferation in INS-1 cells (Table 2). No exogenous stimulus was able to overcome this effect, e.g. neither glucose concentration nor any combination with prolactin and IGF-I. These results support our previous observations of irreversible orthovanadate-dependent inhibition of cell proliferation by other growth factors such as GH or HGF in INS-1 cells [6, 7, 11].
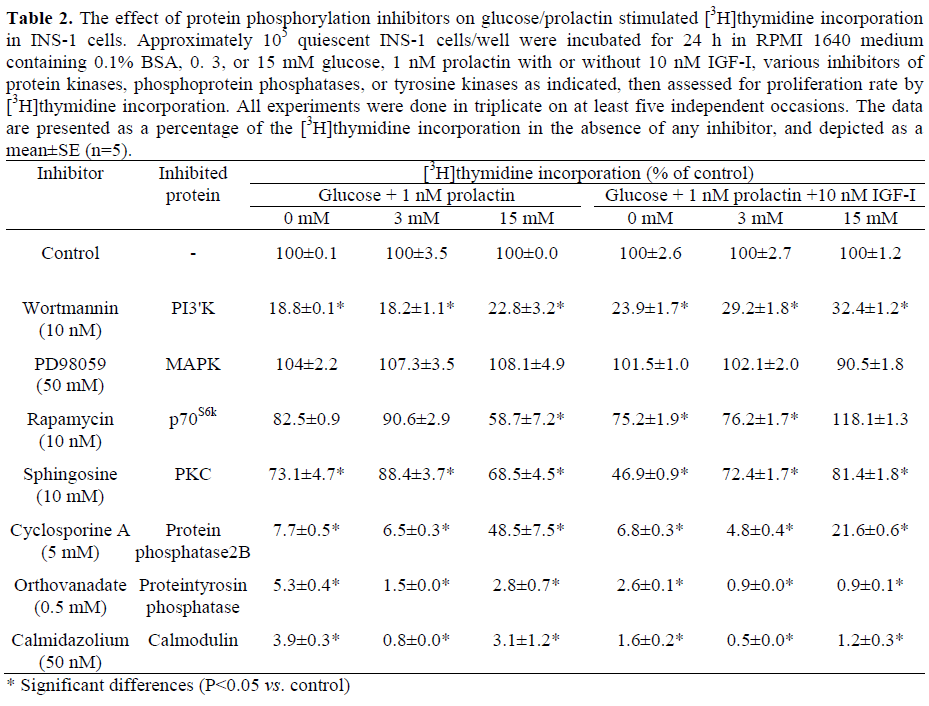
[3H]thymidine incorporation in INS-1 cells in the presence of 5μM cyclosporine A, an inhibitor of protein phosphatase 2B [25] largely abolished prolactin-stimulated cell proliferation (reduction greater than 90%, Table 2). However, in the presence of high glucose concentrations (15 mM), the inhibition by cyclosporine A was significantly reversed (52% reduction at 15 mM glucose plus 1 nM prolactin, P=0.012).
Inhibition of PI3’K activity using 10 nM wortmannin [26] significantly reduced prolactin stimulated INS-1 cell proliferation by 77% at 15 mM glucose and by 82% at 3 mM glucose (P<0.001, Table 2). Additional stimulation with IGF-I slightly reversed the reduction of [3H]thymidine incorporation (71% and 68% reduction at 15 mM and 3 mM glucose, respectively), but this effect was not significant (P=0.800 and P=0.065, respectively).
p70S6k is activated downstream of PI3’K [27]. Rapamycin (10 nM), a specific inhibitor of p70S6k, reduced [3H]thymidine incorporation in prolactin-stimulated INS-1 cells by 42% in the presence of high glucose concentrations (15 mM) but only by 10 to 18% in the absence of glucose or at low glucose levels which was not significant (P=0.365 at 3 mM, P=0.115 at 0 mM glucose; Table 2). After stimulation with prolactin and IGF-I, there was an even stronger difference between low glucose concentrations, which showed a 25% reduction, and high glucose concentrations, where no inhibition by rapamycin, but only a slight stimulation of cell proliferation was seen (Table 2). This effect was not observed after stimulation with other growth factors [6, 7, 11] and suggests that prolactin and IGF-I activate alternative pathways which are not p70S6k-dependent.
Inhibition of protein kinase C (PKC) by sphingosine (10 μM) [28] resulted only in a moderate reduction of prolactin-stimulated [3H]thymidine incorporation (31% in the presence of 15 mM glucose, P<0.001); additional stimulation with IGF-I was able to partially overcome this effect (19% reduction). Still, inhibition of INS-1 cell proliferation remained significant (P=0.012). These findings suggest that certain PKC isoforms might be involved in the regulation of prolactin-mediated INS-1 cell proliferation even if they do not seem to be of major importance, especially in the presence of IGF-I. Inhibition of the MAPK kinase (MEK), the activator of MAPK, by PD98059 (50 μM) [29] did not change the prolactin-dependent proliferation rate of INS-1 cells significantly either in the presence of low (P=0.497) or high glucose concentrations (P=0.373), or in the presence of IGF-I (P values ranging from 0.089 and 0.605). This suggests that the MAPK pathway may not play an important role in prolactin-stimulated INS-1 cell proliferation. High intracellular Ca2+ concentrations are essential for cell proliferation as has been shown before [14, 30]. In our experiments, we used calmidazolium (50 nM) to inhibit Ca2+/calmodulin dependent proteins which reduced [3H]thymidine incorporation in prolactin-stimulated INS-1 cells by more than 95% (P<0.001). This was seen at any glucose concentration, and additional stimulation with IGF-I could not overcome this effect. These data emphasize the essential role of Ca2+/calmodulin for the growth of pancreatic beta-cells (Table 2).
DISCUSSION
Prolactin is known to be one of the most potent growth factors for pancreatic beta-cell proliferation [9, 15, 16, 17, 23, 31]. However, little is known about the molecular mechanisms by which prolactin-dependent pancreatic beta-cell proliferation is induced. In this study, we show that prolactin stimulates proliferation of the glucosedependent beta-cell line INS-1, in particular, in the presence of physiological and subphysiological glucose concentrations (from a 2.7- to a 3.5-fold above stimulation with glucose alone at 3-6 mM; Figure 2). Prolactin shows significant proliferation even in the absence of glucose. This is different from the stimulatory effects seen with other growth factors [6, 7]. Proliferation increased most in the presence of physiological glucose concentrations (6 mM) whereas the strongest additional effect of prolactin above glucose was seen at low glucose concentrations (3 mM). The proliferative effect of prolactin was significantly lower in the presence of high glucose concentrations (2-fold as compared to stimulation with 15 mM glucose alone, P=0.019). A similar effect was previously seen after stimulation with hepatocyte growth factor (HGF) [11] whereas stimulation with IGF-I or GH displayed the maximum stimulatory effect in the presence of 15 mM glucose (about 4-fold as compared to stimulation with glucose alone) [6, 7]. The biological reason for this effect is probably that a greater insulin need and therefore augmented beta-cell proliferation is provided during pregnancy and embryogenesis even if the glucose levels stay in the physiological or even subphysiological range. Thus, the organism uses different tools to react to different situations of beta-cell need. The stimulatory effect of prolactin was as strong as the effect of IGF-I (Figure 3) and surpassed the effect of other growth factors (GH; Figure 3, Table 1; HGF maximum 2.2-fold increase in [3H]thymidine incorporation, [6]). In contrast to the experiments with other growth factors [6, 7, 11, 32], INS-1 cells also showed a marked proliferation in the presence of prolactin even if no glucose was present (about 2.3-fold as compared to unstimulated controls; Figure 1). However, co-stimulation of INS-1 cells with prolactin, glucose and other growth factors did instead decrease [3H]thymidine even though this effect was not significant (Figure 3, Table 1). This suggests that there may be a saturation effect after stimulation of INS-1 cells with growth factors, and that growth factors may use a limited number of signal transduction pathways in common. It might also be that extensive proliferation leads to activation of cellular factors which inhibit cell proliferation. Therefore, stimulation with additional growth factors may not necessarily result in increased proliferation of pancreatic beta-cells, at least, when using the INS-1 cell line. Here as well, the cells and probably the organism avoid unlimited cell proliferation. Further experiments are required before extrapolating the results to primary cells.
Prolactin induced beta-cell proliferation was highly dependent on PI3’K activation; inhibition of PI3’K activity by the specific inhibitor wortmannin largely abolished INS-1 cell proliferation regardless of the stimulus used in our experiments (Table 2). These results underscore the central role of PI3’K in growth factor dependent pancreatic beta-cell proliferation since we have previously shown that the effects of other hormonal ligands are dependent on PI3’K as well [6, 7, 11]. PI3’K activation by different growth factors occurs via differing signal transduction pathways [23, 33, 34, 35]. IGF-I primarily activates PI3’K via IRS-2 [7]. In contrast, prolactin triggers glucose-dependent different signaling protein activation; in the presence of low glucose concentrations (3 mM), prolactin mainly activates mainly IRS-2 (Figure 5). The other members of the IRS-family are less activated. IRS-4 shows its strongest activation at physiological glucose concentrations (6 mM) (Figure 6) and IRS-1 in the presence of high glucose concentrations (15 mM) (Figure 4). This suggests that hormonal effects are much more complex and that proteins like prolactin activate different signal transduction pathways dependent on interaction with different nutrients or growth factors.
Activated IRS-2 and IRS-4 seem to have the strongest impact on cell proliferation since maximal proliferation of prolactin-stimulated INS-1 cells occurred in the presence of 3-6 mM glucose (Figure 2). This is congruent with data which show that IRS-2 overexpression increases beta-cell proliferation much more than other IRSproteins and that IRS-4 overexpression is capable of partially compensating for an IRS- 2 decrease [32, 36, 37].
Prolactin also stimulates proliferation in the presence of physiological glucose concentration via the JAK-2/STAT-5 pathway (Figure 7) as do HGF and GH [6, 11]. But this pathway seems to be of lesser importance at low and high glucose concentrations since the JAK-2/STAT-5 signal transduction pathway is less activated under these conditions (Figure 7).
The prolactin-activated signal transduction pathways split downstream of PI3’K (Figure 8) as was seen after stimulation with other growth factors [6, 7, 11]. Activation of p70s6k and PKC seems to play a role in the presence of high glucose concentrations as is suggested by protein inhibitor experiments. Inhibition of p70s6k by rapamycin reduced prolactinstimulated INS-1 cell proliferation moderately but significantly (Table 2) whereas p70s6k seems to be of lesser importance in the presence of low glucose concentrations. A similar pattern was seen with specific inhibition of PKC by sphingosine. Further experiments are required to clarify which pathways are activated downstream of PI3’K in the presence of low glucose.
Figure 8. Prolactin-dependent signal transduction
pathways. Prolactin-activated beta-cell signalling in the
presence of physiological glucose concentration (6
mM). Glc: glucose; Grb2: growth factor bound protein
2; JAK-2: janus kinase 2; MAPK: mitogen activated
protein kinase; MEK: MAPK kinase; IGF-I: insulin
like growth factor-I; IRS: insulin receptor substrate;
PI3‘K: phosphatidylinositol 3‘ kinase; PKB: protein
kinase B = AKT; PKC: protein kinase C; PKG: protein
kinase G; Prl: prolactin; p70S6k: 70kD S6 kinase; Shc:
SH2-containing protein; mSOS: murine sons of
sevenless-1 protein; STAT-5: signal transducer and
activator of transcription; mTOR: murine target of
rapamycin.
The MAPK pathway seems to be of little significance in prolactin-mediated INS-1 cell proliferation. Inhibition of MAPK activity by PD98059 showed no significant decrease in prolactin-stimulated beta-cell proliferation (Table 2) and no change of MAPK phosphorylation; subsequent activation was seen after prolactin stimulation (data not shown). Neither did the signal transduction proteins, murine sons of sevenless-1 protein (mSOS) and MEK upstream of MAPK, display any change of activation upon prolactin stimulation in INS-1 cells (data not shown). GH- and HGF-stimulated INS-1 cell proliferation also does not involve the MAPK pathway, as we have demonstrated before [6, 11]. In contrast, INS-1 cell proliferation stimulated by IGF-I does involve MAPK activation [12]. However, the activation of Shc is seen in the presence of all glucose concentrations after stimulation with prolactin (data not shown) which suggests the activation of further pathways downstream of Shc.
Another essential condition in growth factor and glucose-mediated pancreatic beta-cell proliferation is the intracellular Ca2+ level. Inhibition of calmodulin kinase by calmidazolium leads to an almost complete inhibition of prolactin stimulated INS-1 cell proliferation (more than 95%, P<0.001; Table 2). A comparable result has been observed after stimulation with HGF [11]. In contrast, IGF-I- or GH- stimulated cell proliferation showed no significant decrease in the presence of calmidazolium [7]. It is interesting that prolactin is therefore able to inhibit the effect seen after stimulation with IGF-I since IGF-I is not able to overcome the inhibitory effect of calmidazolium in the presence of prolactin (Table 2). These findings provide evidence that calmodulindependent proteins are of importance only for prolactin- and HGF-stimulated INS-1 cell proliferation and that prolactin also seems to activate inhibitory pathways especially in the presence of high glucose concentrations which have to be analyzed in further experiments.
In summary, this study provides evidence that prolactin triggers cell proliferation glucose dependently via various signal transduction pathways with a maximum stimulatory effect in the presence of physiological glucose concentrations. Prolactin-induced INS-1-cell proliferation is accompanied by activation of IRS-2, IRS-4 and JAK-2/STAT-5 proteins, and subsequent PI3’K activation. These effects are glucose-dependent. Co-stimulation with other growth factors showed no synergistic effect while stimulation by either IGF-I or GH can be enhanced in combination with additional growth factors. The MAPK pathway seems to be of minor importance for prolactin-stimulated beta-cell proliferation. Future studies are necessary to identify the respective transcription factors which enable crosstalk between the various pathways and the signal transduction proteins which are activated downstream of PI3’K, PKC and p70S6k.
Acknowledgment
This work was supported by a grant (Hu765/2-1) of the German Research Society (DFG)
Conflict of interest
The authors have no potential conflicts of interest
References
- Brockenbrough JS, Weir GC, Bonner-Weir S. Discordance of exocrine and endocrine growth after 90% pancreatectomy in rats. Diabetes 1988; 37:232-6. [PMID 3292318]
- Swenne I. Pancreatic beta-cell growth and diabetes mellitus. Diabetologia 1992; 35:193-201. [PMID 1563578]
- Newgard CB, McGarry JD. Metabolic coupling factors in pancreatic beta-cell signal transduction. Annu Rev Biochem 1995; 64:689-719. [PMID 7574498]
- Prenki M. New insights into pancreatic beta-cell metabolic signaling in insulin secretion. Eur J Endocrinol 1996; 134:272-86. [PMID 8616523]
- Chick WL. Beta cell replication in rat pancreatic monolayer cultures. Effects of glucose, tolbutamide, glucocorticoid, growth hormone and glucagon. Diabetes 1973; 22:687-93. [PMID 4125576]
- Cousin SP, Hügl SR, Myers MG Jr, White MF, Reifel-Miller A, Rhodes CJ. Stimulation of pancreatic beta-cell proliferation by growth hormone is glucosedependent: signal transduction via Janus kinase 2 (JAK2)/signal transducer and activator of transcription 5 (STAT5) with no crosstalk to insulin receptor substrate-mediated mitogenic signalling. Biochem J 1999; 344:649-658. [PMID 10585851]
- Hügl SR, White MF, Rhodes CJ. Insulin-like growth factor I (IGF-I)-stimulated pancreatic beta-cell growth is glucose-dependent. Synergistic activation of insulin receptor substrate-mediated signal transduction pathways by glucose and IGF-I in INS-1 cells. J Biol Chem 1998; 273:17771-9. [PMID 9651378]
- Cousin SP, Hügl SR, Wrede CE, Kajio H, Myers MG, Jr.Rhodes CJ. Free fatty acid-induced inhibition of glucose and insulin-like growth factor I-induced deoxyribonucleic acid synthesis in the pancreatic betacell line INS-1. Endocrinology 2001; 142:229-240. [PMID 11145586]
- Billestrup N, Nielsen JH. The stimulatory effect of growth hormone, prolactin, and placental lactogen on beta-cell proliferation is not mediated by insulin-like growth factor-I. Endocrinology 1991; 129:883-8. [PMID 1677331]
- Nielsen JH, Linde S, Welinder BS, Billestrup N, Madsen OD. Growth hormone is a growth factor for the differentiated pancreatic beta-cell. Mol Endocrinol 1989; 3:165-73. [PMID 2644530]
- Gahr S, Merger M, Bollheimer LC, Hammerschmied CG, Schölmerich J, Hügl SR. Hepatocyte growth factor stimulates proliferation of pancreatic beta-cells particularly in the presence of subphysiological glucose concentrations. J Mol Endocrinol 2002; 28:99-110. [PMID 11932207]
- Dickson LM, Lingohr MK, McCuaig J, Hügl SR, Snow L, Kahn BB, et al. Differential activation of protein kinase B and p70(S6)K by glucose and insulinlike growth factor 1 in pancreatic beta-cells (INS-1). J Biol Chem 2001; 276:21110-20. [PMID 11274216]
- Welsh M, Welsh N, Nilsson T, Arkhammar P, Pepinsky RB, Steiner DF, Berggren PO. Stimulation of pancreatic islet beta-cell replication by oncogenes. Proc Natl Acad Sci U S A 1988; 85:116-20. [PMID 2829167]
- Frödin M, Sekine N, Roche E, Filloux C, Prentki M, Wollheim CB, Van Obberghen E. Glucose, other secretagogues, and nerve growth factor stimulate mitogen- activated protein kinase in the insulinsecreting beta-cell line, INS-1. J Biol Chem 1995; 270:7882-9. [PMID 7713882]
- Labriola L, Montor WR, Krogh K, Lojudice FH, Genzini T, Goldberg AC, et al. Beneficial effects of prolactin and laminin on human pancreatic islet-cell cultures. Mol Cell Endocrinol 2007; 263:120-33. [PMID 17081683]
- Nielsen JH, Moldrup A, Billestrup N, Petersen ED, Allevato G, Stahl M. The role of growth hormone and prolactin in beta cell growth and regeneration. Adv Exp Med Biol 1992; 321:9-17. [PMID 1449086]
- Brelje TC, Parsons JA, Sorenson RL. Regulation of islet beta-cell proliferation by prolactin in rat islets. Diabetes 1994; 43:263-73. [PMID 7904577]
- Galsgaard ED, Friedrichsen BN, Nielsen JH, Moldrup A. Expression of dominant-negative STAT5 inhibits growth hormone- and prolactin-induced proliferation of insulin-producing cells. Diabetes 2001; 50 Suppl 1:S40-1. [PMID 11272198]
- Asfari M, Janjic D, Meda P, Li G, Halban PA, Wollheim CB. Establishment of 2-mercaptoethanoldependent differentiated insulin-secreting cell lines. Endocrinology 1992; 130:167-78. [PMID 1370150]
- Hofmann C, White MF, Whittaker J. Human insulin receptors expressed in insulin-insensitive mouse fibroblasts couple with extant cellular effector systems to confer insulin sensitivity and responsiveness. Endocrinology 1989; 124:257-64. [PMID 2462489]
- Cheatham B, Vlahos CJ, Cheatham L, Wang L, Blenis J, Kahn CR. Phosphatidylinositol 3-kinase activation is required for insulin stimulation of pp70 S6 kinase, DNA synthesis, and glucose transporter translocation. Mol Cell Biol 1994; 14:4902-11. [PMID 8007986]
- Myers MG, Jr., Grammer TC, Wang LM, Sun XJ, Pierce JH, Blenis J, et al. Insulin receptor substrate-1 mediates phosphatidylinositol 3'-kinase and p70S6k signaling during insulin, insulin-like growth factor-1, and interleukin-4 stimulation. J Biol Chem 1994; 269:28783-9. [PMID 7961833]
- Friedrichsen BN, Galsgaard ED, Nielsen JH, Moldrup A. Growth hormone- and prolactin-induced proliferation of insulinoma cells, INS-1, depends on activation of STAT5 (signal transducer and activator of transcription 5). Mol Endocrinol 2001; 15:136-48. [PMID 11145745]
- Galsgaard ED, Nielsen JH, Moldrup A. Regulation of prolactin receptor (PRLR) gene expression in insulin-producing cells. Prolactin and growth hormone activate one of the rat prlr gene promoters via STAT5a and STAT5b. J Biol Chem 1999; 274:18686-92. [PMID 10373481]
- Groblewski GE, Wagner AC, Williams JA. Cyclosporin A inhibits Ca2+/calmodulin-dependent protein phosphatase and secretion in pancreatic acinar cells. J Biol Chem 1994; 269:15111-7. [PMID 7515049]
- Ogreid D, Dostmann W, Genieser HG, Niemann P, Doskeland SO, Jastorff B. (Rp)- and (Sp)-8- piperidino-adenosine 3',5'-(cyclic)thiophosphates discriminate completely between site A and B of the regulatory subunits of cAMP-dependent protein kinase type I and II. Eur J Biochem 1994; 221:1089-94. [PMID 8181466]
- Kardalinou E, Zhelev N, Hazzalin CA, Mahadevan LC. Anisomycin and rapamycin define an area upstream of p70/85S6k containing a bifurcation to histone H3-HMG-like protein phosphorylation and cfos- c-jun induction. Mol Cell Biol 1994; 14:1066-74. [PMID 8289787]
- Liu JP. Protein kinase C and its substrates. Mol Cell Endocrinol 1996; 116:1-29. [PMID 8822261]
- Pang L, Sawada T, Decker SJ, Saltiel AR. Inhibition of MAP kinase kinase blocks the differentiation of PC-12 cells induced by nerve growth factor. J Biol Chem 1995; 270:13585-8. [PMID 7775407]
- Khoo S, Cobb MH. Activation of mitogenactivating protein kinase by glucose is not required for insulin secretion. Proc Natl Acad Sci U S A 1997; 94:5599-604. [PMID 9159118]
- Brelje TC, Sorenson RL. Role of prolactin versus growth hormone on islet B-cell proliferation in vitro: implications for pregnancy. Endocrinology 1991; 128:45-57. [PMID 1986937]
- Lingohr MK, Dickson LM, McCuaig JF, Hügl SR, Twardzik DR, Rhodes CJ. Activation of IRS-2- mediated signal transduction by IGF-1, but not TGFalpha or EGF, augments pancreatic beta-cell proliferation. Diabetes 2002; 51:966-76. [PMID 11916914]
- Krasilnikov MA. Phosphatidylinositol-3 kinase dependent pathways: the role in control of cell growth, survival, and malignant transformation. Biochemistry 2(mosc) 2000; 65:59-67. [PMID 10702641]
- Reddy EP, Korapati A, Chaturvedi P, Rane S. IL-3 signaling and the role of Src kinases, JAKs and STATs: a covert liaison unveiled. Oncogene 2000; 19:2532-47. [PMID 10851052]
- Vanhaesebroeck B, Alessi DR. The PI3K-PDK1 connection: more than just a road to PKB. J Biochem 2000; 346(Pt 3):561-76. [PMID 10698680]
- Lingohr MK, Dickson LM, Wrede CE, Briaud I, McCuaig JF, Myers MG, Jr, Rhodes CJ. Decreasing IRS-2 expression in pancreatic beta-cells (INS-1) promotes apoptosis, which can be compensated for by introduction of IRS-4 expression. Mol Cell Endocrinol 2003; 209:17-31. [PMID 14604813]
- Lingohr MK, Dickson LM, Wrede CE, McCuaig JF, Myers MG Jr, Rhodes CJ. IRS-3 inhibits IRS-2- mediated signaling in pancreatic beta-cells. Mol Cell Endocrinol 2003; 204:85-99. [PMID 12850284]









