Keywords
Prognosis; Survival
Abbreviations
neuroendocrine carcinoma; PNET Pancreatic neuroendocrine tumors
INTRODUCTION
Pancreatic neuroendocrine tumors (PNET) are rare [1], with an incidence of less than one per 100000 populations per year. The first description of this type of tumors was published by Oberndorfer in 1907 [2]. In the past various terms have been used for this type of lesion, including: carcinoid tumor, endocrine tumor, neuroendocrine carcinoma, apudoma, and gastroenteropancreatic tumor. In 2000, the World Health Organization (WHO) introduced a structured classification of gastroenteropancreatic neuroendocrine tumors and subdivided them into three main categories: well differentiated neuroendocrine tumor (NET), well differentiated neuroendocrine carcinoma (NEC), and poorly differentiated NEC [3, 4].
In 2010 a revised version of the WHO classification of PNET was published [5]. The term “neuroendocrine” refers to neoplastic cells, which are characterized by the expression of molecular differentiation markers such as e.g. chromogranin A, synaptophysin and specific enolase. These markers are of diagnostic importance for this tumor entity and can be proved immunohistochemically. The term “neuroendocrine tumor” (NET) includes all well and poorly differentiated neoplasms of neuroendocrine cells. The WHO classification classifies NET based on the Ki-67 index and the evaluation of mitoses in histological material. NET of the gastrointestinal tract and pancreas are differentiated in two groups: Well differentiated NET and poorly differentiated NEC (Table 1). Well differentiated NET are divided depending on their proliferative activity into either G1 (mitotic count of < 2 per 10 high power fields [HPF]: HPF = 2 cm2, 40× magnification and/or ≤ 2% Ki-67 index) and G2 tumors (mitotic count of 2–20 per HPF and/ or 3–20% Ki-67 index). Poorly differentiated NEC, which are G3 tumors (mitotic count of >20 per 2 HPF and/or >20% Ki-67 index), are subtyped into small cell neoplasms, large cell neoplasms and mixed adenoneuroendocrine carcinoma (MANEC) [7]. This classification system founds on the grading system formerly proposed by the European Neuroendocrine Tumor Society (ENETS) [5-7]. The WHO- 2010 staging system can effectively evaluate the prognosis.

NET represent only 1–3% of all pancreatic neoplasms, but the incidence has increased two- to threefold in recent decades [8, 9]. Classification into functional and nonfunctional tumors also depends on the associated clinical syndromes [10, 11]. Functional tumors secrete biologically active peptides such as insulin, gastrin, glucagon, somatostatin, and vasoactive intestinal polypeptides [11]. Nonfunctional tumors are usually identified incidentally-for example, when they became symptomatic due to tumor bulk, or when imaging procedures are being carried out for other indications.
Most of the lesions are sporadic, or they may be part of a genetic syndrome such as multiple endocrine neoplasia type 1 (MEN1), von Hippel–Lindau syndrome, neurofibromatosis type 1, or tuberous sclerosis [12].
In general, complete surgical resection of the tumor should be attempted [13, 14]. However, for nonfunctional tumors the malignant potential seems diverse. Small symptomatic nonfunctional tumors often seem to have benign course, thus these patients may be candidates for surveillance. However, there exist no clear cut-off criteria for size to distinguish between benign and malignant disease [12]. In particular large nonfunctional tumors are associated with worse outcome [13]. The prognosis after surgical resection is excellent; the long-term survival for patients with resected insulinomas is more than 90%, for example [14]. In contrast to patients with pancreatic adenocarcinoma, hepatic resection and palliative resection of the primary tumor may be beneficial and is associated with significant survival benefit in patients with metastatic PNET [16, 17]. If the majority of the tumor can be resected, debulking surgery may be justified [14]. Recent studies evaluate the role of long-acting somatostatin analogues (S-LAR), which appear to have a significant antiproliferative effect and improve long-term survival of selected patients with metastatic NET when administration of S-LAR is combined with aggressive cytoreductive Surgery [18].
A retrospective analysis of patients treated at the University of Bochum was carried out in order to evaluate experience with these neuroendocrine tumors in our own institution. Short-term and long-term outcomes were analyzed, as well as prognostic factors influencing survival. The influence of functional and nonfunctional PNET’s on long-term survival was also assessed.
MATERIALS AND METHODS
The clinical data for 49 patients with neuroendocrine pancreatic and peripancreatic neuroendocrine tumors, who had been operated from January 2004 to December 2010 were analyzed retrospectively. Patients were informed about this high-class security study when the operation method was explained before surgery.
All patients had histopathologic confirmation of an endocrine tumor. The review included the patients’ demographic data, surgical data, postoperative morbidity and mortality, long-term survival, and prognostic factors. Also the following features were evaluated: grading (according to the WHO-2010 criteria), staging (according to the AJCC/UICC 2009 criteria for well differentiated tumors), distant and locoregional lymph node metastasis at diagnosis, hormonal activity, main symptoms, and simultaneous presence of other neoplasms.
All patients were followed up, and the long-term overall survival for each patient was calculated from the date of surgery to the date of death or last follow-up. The end of the follow-up period for surviving patients was March 2011. Neuroendocrine tumors associated with the relevant symptoms, signs, and laboratory evidence of hormonal excess were regarded as functioning tumors and were further classified according to the syndromes present. Postoperative mortality was defined as death occurring within the first 30 postoperative days.
Statistical Analysis
Statistical analysis was carried out using the IBM SPSS 20 software package (IBM Corporation, Armonk, New York, USA). Overall survival was calculated from the date of resection to the date of death. Survival data were obtained from the hospital database. Cumulative survival data were calculated using the Kaplan–Meier method, and significance was assessed using log-rank (Mantel– Cox) testing. A univariate survival analysis was carried out for continuous data using Cox proportional hazards regression. Multivariate survival analysis was conducted using Cox regression. Significance was set at P<0.05.
RESULTS
A total of 49 patients underwent surgery for neuroendocrine tumors in the pancreas (n=43; 87.8%) or peripancreatic region from January 2004 to December 2010. There were 31 men (63.3%) and 18 women (36.7%). The patients’ median age at diagnosis was 59 years (range 17–83 years). The tumor was located in the head of the pancreas in 13 patients (26.5%), in the body of the pancreas in three (6.1%), in the pancreatic tail in 27 (55.1%), and in the peripancreatic region in six patients (12.2%), including the ampulla of Vater (n=4; 8.2%) and duodenum (n=2; 4.1%).
The median follow-up period was 29 months (range 1–77 months). Forty patients (81%) had non-functional tumors, nine patients (19%) had functioning tumors (six insulinomas, two gastrinomas, and one somatostatinoma), and two of the patients had MEN1. There was no significant difference in the median age of the patients in the functional and nonfunctional groups.
There were 20 well differentiated G1 tumors (mitotic count of < 2 per HPF and/or ≤ 2% Ki-67 index), 28 well differentiated G2 tumors (mitotic count of 2–20 per HPF and/ or 3–20% Ki-67 index), and only 1 poorly differentiated G3 (mitotic count of >20 per 2 HPF and/or >20% Ki-67 index) MANEC. Tumor sizes ranged from 2 mm to 123 mm, with a median of 27 mm. There was a significant difference in the median tumor size between the functional and nonfunctional groups (13 mm vs. 30 mm; P=0.001). Both functional and nonfunctional tumors were most frequently located in the pancreatic tail. Ten patients (20.4%) had hepatic metastases at diagnosis and only two more patients developed metastases during the follow-up period. Liver metastases were resected in a synchronous approach without any prior therapy. The average survival time of these patients was 30 months.
Six of the abovementioned patients with hepatic metastasis underwent aggressive cytoreductive surgery and additionally were treated with S-LAR and provide an average survival time of 29 months. In two patients with advanced tumors and high proliferation rate adjuvant chemotherapy was performed. Patients are deceased after 19 and 66 months. Involvement of lymph nodes was found in 17 patients (37.7%). Lymph-node metastasis was more common in the nonfunctional group (55% vs. 22%), but the difference was not significant (P=0.058) (Table 1).
Pylorus-preserving or classical pancreaticoduodenectomy was carried out in 14 patients (28.5%), distal pancreatectomy in 27 (55%) patients, total pancreatectomy in four (8%) patients, and segmental pancreas resection or enucleation in four patients (8%). Reasons for total pancreatectomy were the following: 2x extensive tumor size, 1xMEN patient with one neuroendocrine tumor in the head and another neuroendocrine tumor in the tail of the pancreas, 1x lipomatosis of the pancreas.
Excluding one patient who died due to bleeding in the postoperative period, the mean survival period in the 48 patients was 29.5±2.8 months (range 2–77 months). The 1-year, 2-year, and 5-year survival rates were 98.5% (48 patients), 95% (47 patients) and 85.5% (46 patients), respectively (Figure 1).
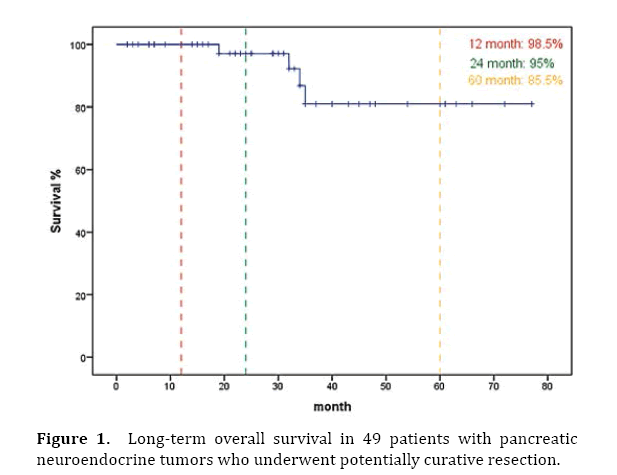
Figure 1. Long-term overall survival in 49 patients with pancreatic neuroendocrine tumors who underwent potentially curative resection.
Forty-four patients (89.8%) were still alive at the time of the analysis. One patient died in the postoperative period, three patients died of tumor progression, and one patient died of a pneumonia and an acute renal failure 52 days after the operation.
Two patients who died of tumor progression had nonresectable liver metastasis. The tumor grade based on WHO criteria was pT1 N0 M1hep; Ki-67 4% respectively pT3 N0 M1 hep; Ki-67 10%. Resection of the pancreatic tumor and atypical liver resection was performed in order to provide tumor debulking. The third patient developed liver metastasis three months after R0 resection was done. Chemotherapy was initiated, however the patient died of tumor progression 32 months after the operation.
It would be interesting to know some details about the three patients who died from tumor progression. Was one of the patients who progressed the patient with poorly differentiated carcinoma? Did the patients who died from progression have residual disease after resection (ie was some of the disease unresectable and left in the patient) or were they disease-free after resection?
Prognostic factors influencing survival were evaluated in a univariate analysis. Significant factors identified were tumor stage (P<0.001), T classification, pathological classification of 2010 (P=0.03), presence of liver metastases (P=0.003), and the resection margin (Figures 2–6). The functional status of the tumor (P=0.164), tumor size (P=0.196), and lymph-node stage (P=0.88) were not found to be significant prognostic factors for survival.
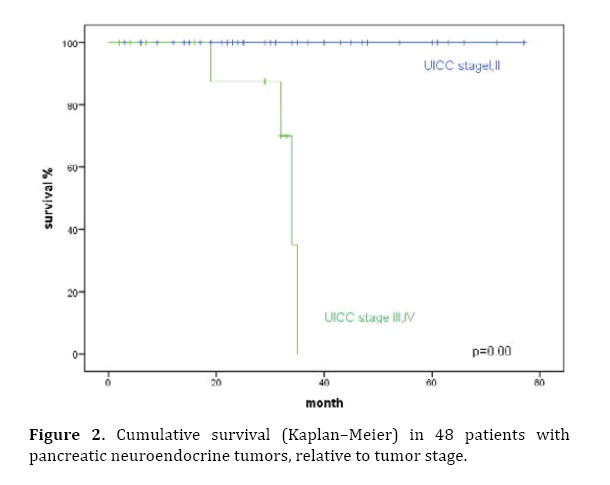
Figure 2. Cumulative survival (Kaplan–Meier) in 48 patients with pancreatic neuroendocrine tumors, relative to tumor stage.
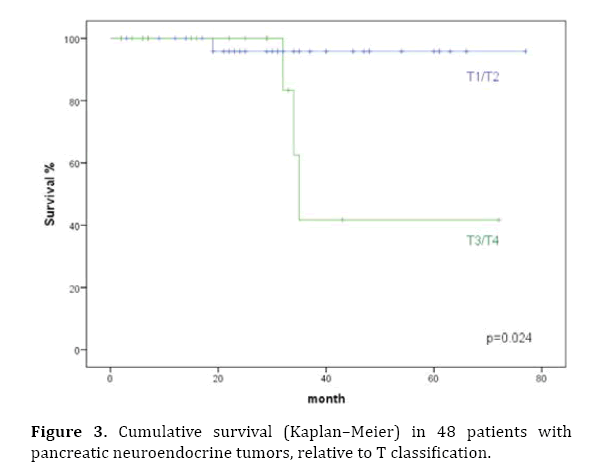
Figure 3. Cumulative survival (Kaplan–Meier) in 48 patients with pancreatic neuroendocrine tumors, relative to T classification.
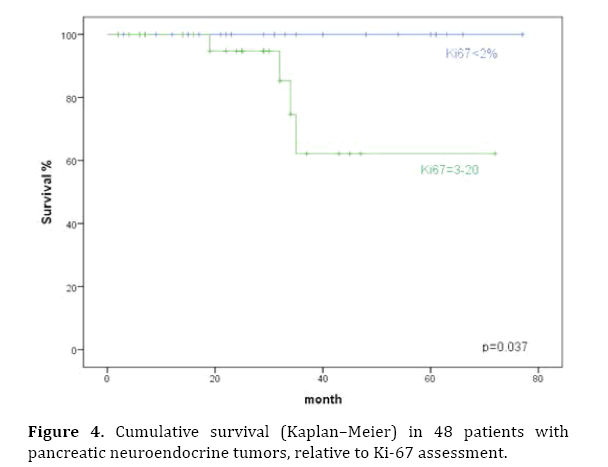
Figure 4. Cumulative survival (Kaplan–Meier) in 48 patients with pancreatic neuroendocrine tumors, relative to Ki-67 assessment.
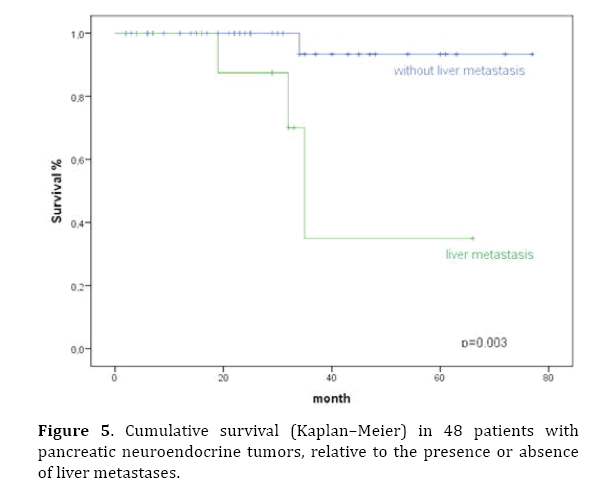
Figure 5. Cumulative survival (Kaplan–Meier) in 48 patients with pancreatic neuroendocrine tumors, relative to the presence or absence of liver metastases.

Figure 6. Cumulative survival (Kaplan–Meier) in 48 patients with pancreatic neuroendocrine tumors, relative to the resection margin (R0 vs. R2).
In the multivariate analysis, tumor stage (HR 72.7; P=0.02), liver metastases (HR 11.6; P=0.038), and the resection margin (HR 0.068; P=0.022) were found to be independent prognostic factors (Table 2).

DISCUSSION
The biological behavior of PNET is less aggressive than that of pancreatic adenocarcinomas, even in the metastatic stage [1, 19, 20, 21]. As a result of improved diagnostic methods and a higher rate of resection, the incidence of PNET has been increasing [4, 22]. Various studies have reported 5-year survival rates for PNET resection ranging from 59% to 80% [22, 23]. In our study the 5-year survival rate was 85.5%. According to the literature almost half of PNET are non-functioning [24].
Surgical resection is the only potentially curative treatment available for the disease and has been reported to decrease the risk of future metastasis and disease-specific mortality [13, 20, 25; 26]. Pancreatic resection results in greater than 80% 3-year survival in non-metastatic PNET.
In this study the ratio of non-functioning tumor is high (81%) compared with previously reported series which showed that half of PNET are non-functioning [24]. The great majority of resected non-functioning tumors was >2 cm (median: 3 cm; range 0.2-12.3 cm). Only in small (≤ 2 cm) non-functioning PNET the role of surgery is still a matter of question as the these tumors often are benign [24; 26; 27, 28, 29]. However, in our opinion a wait and see policy in these patients is critical as there exists potential malignancy and a risk of development of metastasis even in small non-functioning PNET. Thus, our strategy of a surgical approach with parenchyma-sparing surgery even in small non-functioning NET is in line with the consideration of other recent publications [13, 28].
Even in metastatic disease data suggest that aggressive surgical management of hepatic metastases improve survival [30]. However, patients with more than 50% liver involvement may not benefit from an aggressive approach [17]. In patients with metastatic neuroendocrine tumors application of S-LAR combined with aggressive cytoreductive surgery and a multidisciplinary multimodal approach (liver ablation, hepatic chemoembolization systemic treatments) further improve survival [17, 18, 30]. In our study 6 patients with metastatic neuroendocrine tumor underwent aggressive cytoreductive surgery plus S-LAR due to incomplete resection of metastasis. All patients survived until now (median 29 months) maybe due to a low proliferation index and low tumor load.
Various surgical procedures were carried out in the present study. The overall complication rate was 26.5% and the perioperative mortality rate was 2%. These results are in accordance with those from other centers [1, 17]. The male–female ratio showed a predominance of men in the present study group (63.3% vs. 36.7%), whereas Gullo et al. [31] and Tomassetti et al. [32] reported a predominance of women. The average age at the time of diagnosis was 56 years, similar to that reported in previous studies [32]. The percentage of patients with non-functional tumors of the pancreas was 81%. A greater incidence of nonfunctional tumors was also observed in several recent studies [33, 34]. As most nonfunctional tumors are asymptomatic, they are usually diagnosed incidentally (89% in the present study), due to improved preoperative diagnosis. The present study confirms that functional tumors are smaller in comparison with nonfunctional ones (1.3 cm vs. 3.7 cm) [35]. Both types of tumor are more often located in the pancreatic tail (non-functional 60%, functional 66.7%). An increased incidence of neuroendocrine tumors in the pancreatic head has been reported [20, 34, 35]. The 5-year survival rate in the present study was 85.5%. This is in strong contrast to the outcomes for patients with PNET described in the literature. Wang et al. [36] reported a 5-year survival rate of 65.6% for patients with unresectable tumors. Phan et al. [35] described survival rates of 77% in patients with functional tumors and 52% in those with nonfunctional tumors. However, other studies have reported no differences in the survival rates between the two groups [11, 18]. Several studies have reported that the long-term survival appears to be associated with following prognostic factors: age, sex, functional status of the tumor, tumor grade and TNM stage, pancreatic resection, surgical margin, tumor size and location, and lymph-node or other distant metastases [1, 23, 32, 37]. The present study also confirms that tumor stage (P=0.00), pathological classification (P=0.03), presence of hepatic metastasis (P=0.003), and resection margin are prognostic factors. However lymph node metastasis was more common (45% vs. 22%) and median tumor size was lager (3 vs. 1.3 cm) in non-functional tumors, our study found that functional status was not a significant prognostic factor in the univariate analysis. In contrast, several series have reported a lower 5-year survival rate in patients with non-functional lesions in comparison with those with functioning PNET [35, 38]. Although not significant the in our study observed trend towards lymph node metastasis and increased tumor size may reflect a more aggressive character of non-functional tumors. The absence of a significant effect on survival rate has also been reported by Deutsch et al. [18]. In addition, nonfunctioning tumors often remain asymptomatic and usually are detected at a later stage (larger in size and more often lymph node metastasis) in comparison to functioning PNET that develop symptoms following production of hormones even if the tumor is small of size.
CONCLUSIONS
In conclusion, surgical resection should be attempted and should play a central role in the therapeutic approach to patients with neuroendocrine tumors. The important aspect is early diagnosis, which makes it possible to carry out radical surgery before the tumor has metastasized. Further studies with larger numbers of patients are required.
Conflict of interest
All the authors have no conflicts of intrest.
References
- Fischer L, Kleeff J, Esposito I, Hinz U, Zimmermann A, Friess H, Büchler MW. Clinical outcome and long-term survival in 118 consecutive patients with neuroendocrine tumours of the pancreas. Br J Surg 2008; 95:627–635. [PMID: 18306152]
- Oberndorfer S. KarzinoideTumoren des Dünndarms. Frankf Z Pathol 1905; 1:425–432.
- Solcia E, Rindi G, Buffa R, Fiocca R, Capella C. Gastric endocrine cells: types, function and growth. Regul Pept 2000; 93:31–35. [PMID: 11033050]
- Kloppel G, Perren A, Heitz PU. The gastroenteropancreatic neuroendocrine cell system and its tumors: the WHO classification. Ann N Y Acad Sci 2004; 1014:13–27. [PMID: 15153416]
- Kloppel G, Rindi G, Perren A, Komminoth P, Klimstra DS. The ENETS and AJCC/UICC TNM classifications of the neuroendocrine tumors of the gastrointestinal tract and the pancreas: a statement. Virchows Arch 2010; 456:595–7. [PMID: 20422210]
- Rindi G, Kloppel G, Alhman H, Caplin M, Couvelard A, de Herder WW, et al. TNM staging of foregut (neuro)endocrine tumors: a consensus proposal including a grading system. Virchows Arch 2006;449:395–401. [PMID: 16967267]
- Bosman F, Camerio F, Hruban R, Theise N. WHO classification of tumours of the digestive system. Lyon: IARC Press; 2010.
- Bilimoria KY, Tomlinson JS, Merkow RP, Stewart AK, Ko CY, Talamonti MS, Bentrem DJ. Clinicopathologic features and treatment trends of pancreatic neuroendocrine tumors: analysis of 9,821 patients. J Gastrointest Surg 2007; 11:1460–1467. [PMID: 17846854]
- Fitzgerald TL, Hickner ZJ, Schmitz M, JKort EJ. Changing incidence of pancreatic neoplasms: a 16-year review of state wide tumor registry. Pancreas 2008; 37:134-8. [PMID: 18665072]
- Norton JA, Kivlen M, Li M, Schneider D, Chuter T, Jensen RT. Morbidity and mortality of aggressive resection in patients with advanced neuroendocrine tumors. Arch Surg 2003; 138:859-66. [PMID: 12912744]
- Hochwald SN, Weiser MR, Colleoni R, Brennan MF, Conlon KC. Laparoscopy predicts metastatic disease and spares laparotomy in selected patients with pancreatic non-functional islet cell tumors. Ann Surg Oncol 2001; 8:249–253. [PMID: 11314942]
- Ehehalt F, Saeger HD, Schmidt CM, Grützmann R. Neuroendocrine tumors of the pancreas. Oncologist 2009; 14:456–467. [PMID: 19411317]
- Lombardi M, De Lio N, Funel N, Sardella C, Russo D, Urbani C, Rossi G, Campani D, Martino E, Marcocci C, Boggi U, Bogazzi F. Prognostic factors for pancreatic neuroendocrine neoplasms (pNET) and the risk of small non-functioning pNET. J Endocrinol Invest 2015;38:605-13. [PMID: 25501604]
- Service FJ, McMahon MM, O’Brien PC, Ballard DJ. Functioning insulinoma—incidence, recurrence, and long-term survival of patients: a 60-year study. Mayo ClinProc 1991; 66:711–719. [PMID: 1677058]
- Ong SL, Garcea G, Pollard CA, Furness PN, Steward WP, Rajesh A, Spencer L, Lloyd DM, Berry DP, Dennison AR. A fuller understanding of pancreatic neuroendocrine tumors combined with aggressive management improves outcome. Pancreatology 2009; 9:583–600. [PMID: 19657214]
- Sallinen V, Haglund C, Seppänen H. Outcomes of resected nonfunctional pancreatic neuroendocrine tumors: Do size and symptoms matter? Surgery 2015; S0039-6060(15)00365-7. [PMID: 26070847]
- Touzios JG, Kiely JM, Pitt SC, Rilling WS, Quebbeman EJ, Wilson SD, Pitt HA. Neuroendocrine hepatic metastases: does aggressive management improve survival? Ann Surg 2005; 241:776-83. [PMID:15849513]
- Deutsch GB, Lee JH, Bilchik AJ. Long-Term Survival with Long-Acting Somatostatin Analogues Plus Aggressive Cytoreductive Surgery in Patients with Metastatic Neuroendocrine Carcinoma.J Am Coll Surg 2015;221:26-36. [PMID:26027502]
- Dixon E, Pasieka JL. Functioning and non-functional neuroendocrine tumors of the pancreas.Curr Opin Oncol 2007; 19:30–35. [PMID: 17133109]
- Hochwald SN, Zee S, Conlon KC, Colleoni R, Louie O, Brennan MF, Klimstra DS. Prognostic factors in pancreatic endocrine neoplasms: an analysis of 136 cases with a proposal for low-grade and intermediate-grade groups. J Clin Oncol 2002; 20:2633–2642. [PMID: 12039924]
- O’Grady HL, Conlon KC. Pancreatic neuroendocrine tumours. Eur J Surg Oncol 2008; 34:324–332. [PMID: 17967523]
- Bilimoria KY, Talamonti MS, Tomlinson JS, Stewart AK, Winchester DP, Ko CY, Bentrem DJ. Prognostic score predicting survival after resection of pancreatic neuroendocrine tumors: analysis of 3851 patients. Ann Surg 2008; 247:490–500. [PMID: 18376195]
- Casadei R, Ricci C, Pezzilli R, Campana D, Tomassetti P, Calculli L, Santini D, Antonacci N, Minni F. Value of both WHO and TNM classification systems for patients with pancreatic endocrine tumors: results of a single-center series. World J Surg 2009; 33:2458–2463. [PMID: 19655196]
- Ito T, Sasano H, Tanaka M, Osamura RY, Sasaki I, Kimura W, Takano K, Obara T, Ishibashi M, Nakao K, Doi R, Shimatsu A, Nishida T, Komoto I, Hirata Y, Nakamura K, Igarashi H, Jensen RT, Wiedenmann B, Imamura M. Epidemiological study of gastroenteropancreatic neuroendocrine tumors in Japan. J Gastroenterol 2010; 45:234-43. [PMID: 20058030]
- Hill JS, McPhee JT, McDade TP, Zhou Z, Sullivan ME, Whalen GF, Tseng JF. Pancreatic neuroendocrine tumors: the impact of surgical resection on survival. Cancer 2009; 15;115:741-51. [PMID: 19130464]
- Doi R. Determinants of surgical resection for pancreatic neuroendocrine tumors. J Hepatobiliary Pancreat Sci 2015;22:610-7. [PMID: 25773163]
- Bettini R, Partelli S, Boninsegna L, Capelli P, Crippa S, Pederzoli P, Scarpa A, Falconi M. Tumor size correlates with malignancy in nonfunctioning pancreatic endocrine tumor. Surgery 2011;150:75-82. [PMID: 21683859]
- Falconi M, Zerbi A, Crippa S, Balzano G, Boninsegna L, Capitanio V, Bassi C, Di Carlo V, Pederzoli P. Parenchyma-preserving resections for small nonfunctioning pancreatic endocrine tumors. Ann Surg Oncol 2010;17:1621-7. [PMID: 20162460]
- Falconi M, Bartsch DK, Eriksson B, Klöppel G, Lopes JM, O'Connor JM, Salazar R, Taal BG, Vullierme MP, O'Toole D; Barcelona Consensus Conference participants. ENETS Consensus Guidelines for the management of patients with digestive neuroendocrine neoplasms of the digestive system: well-differentiated pancreatic non-functioning tumors. Neuroendocrinology 2012;95:120-34. [PMID: 22261872]
- Nissen NN, Kim AS, Yu R, Wolin EM, Friedman ML, Lo SK, Wachsman AM, Colquhoun SD. Pancreatic neuroendocrine tumors: presentation, management, and outcomes.Am Surg 2009;75:1025-9. [PMID: 19886158]
- Gullo L, Migliori M, Falconi M, Pederzoli P, Bettini R, Casadei R, DelleFave G, Corleto VD, Ceccarelli C, Santini D, Tomassetti P. Non-functional pancreatic endocrine tumors: a multicenter clinical study. Am J Gastroenterol 2003; 98:2435–2439. [PMID: 14638345]
- Tomassetti P, Campana D, Piscitelli L, Casadei R, Santini D, Nori F, Morselli-Labate AM, Pezzilli R, Corinaldesi R. Endocrine pancreatic tumors: factors correlated with survival. Ann Oncol 2005; 16:1806–1810. [PMID: 16085691]
- Kazanjian KK, Reber HA, Hines OJ. Resection of pancreatic neuroendocrine tumors: results of 70 cases. Arch Surg 2006; 141:765–769. [PMID: 16924083]
- Schurr PG, Strate T, Rese K, Kaifi JT, Reichelt U, Petri S, Kleinhans H, Yekebas EF, Izbicki JR. Aggressive surgery improves long-term survival in neuroendocrine pancreatic tumors: an institutional experience. Ann Surg 2007; 245:273–281. [PMID: 17245182]
- Phan GQ, Yeo CJ, Hruban RH, Lillemoe KD, Pitt HA, Cameron JL. Surgical experience with pancreatic and peripancreatic neuroendocrine tumors: review of 125 patients. J Gastrointest Surg1998; 2:472–482. [PMID: 9843608]
- Wang SE, Su CH, Kuo YJ, Shyr YM, Li AF, Chen TH, Wu CW, Lee CH. Comparison of functional and nonfunctional neuroendocrine tumors in the pancreas and peripancreatic region. Pancreas 2011; 40:253–259. [PMID: 20966805]
- Franko J, Feng W, Yip L, Genovese E, Moser AJ. Non-functional neuroendocrine carcinoma of the pancreas: incidence, tumor biology, and outcomes in 2,158 patients. J Gastrointest Surg 2010; 14:541–548. [PMID: 19997980]
- Eriksson B, Skogseid B, Lundqvist G, Wide L, Wilander E, Oberg K. Medical treatment and long-term survival in a prospective study of 84 patients with endocrine pancreatic tumors. Cancer 1990; 65:1883–1890. [PMID: 1695540]

