Keywords
Individualized Medicine; Pancreatic Neoplasms; Pharmacogenetics
Abbreviations
ASCO American Society of Clinical Oncology; FISH: fluorescence in-situ hybridization; HR hazards ratio; KRASwt: KRAS wild type
Introduction
Pancreatic cancer is an aggressive and lethal malignancy that is recognized as the fourth leading cause of cancer-related deaths in United States. According to National Cancer Institute estimates, over 45,000 new cases and 38,460 deaths were reported in 2013 alone [1]. Despite a better understanding of cancer biology, survival rates remain dismal, with an overall 5-year survival rate of 6% [1]. Surgical resection is the only potential curative option for this disease with the median survival after surgery being only 15 months. Gemcitabine, a nucleoside analog used in various carcinomas, remains the cornerstone of therapy for locally advanced and metastatic pancreatic cancer that is not amenable to resection [2]. This underscores the unmet need for better therapeutics for this dreaded malignancy.
What We Knew Before the 2014 ASCO Gastroenterology Cancers Symposium
Pancreatic cancer has a complex biology with a heterogeneous genetic makeup. The cancer arises from the ductal epithelium and progresses from a pre-malignant lesion to an invasive cancer. This evolution is paralleled by sequential and cumulative gene mutations that have been replicated in mice models [3, 4]. These include activation of KRAS2 oncogene, inactivation of tumor-suppressor gene CDKN2A and finally inactivation of tumorsuppressor gene TP53 and SMAD4 (DPC4) [5, 6, 7]. Almost all pancreatic cancers carry a mutation in one or more of these four genes [6]. A comprehensive genetic analysis of 24 pancreatic cancers showed an average of 63 mutations, most commonly point mutations [4]. With completion of the human genome project, it is now possible to identify the additional numerous gene mutations in pancreatic cancers. The vast majority of these mutations have not yet been analyzed [4] and, although the discovery of some genes has provided further insight into the natural history of the disease, this knowledge has not yet translated to better clinical outcomes. As we discover the complex signaling pathways of the disease and associated genomic mutations, more potential targets emerge for early diagnosis and targeted therapeutics (as shown in Figure 1).
Figure 1. Components and pathways of pancreatic cancer. (Reprinted with permission of Massachusetts Medical Society from: Hidalgo M. Pancreatic Cancer. N Engl J Med 2010; 362:1605-7. Copyright © (2010) Massachusetts Medical Society [6]).
What We Have Learned at the 2014 ASCO Gastrointestinal Cancers Symposium
This review article summarizes the recent work presented at the 2014 ASCO Gastrointestinal Cancers Symposium on genome mutations in pancreatic cancer and pharmacogenomics of the disease. We discuss two abstracts in particular that are retrospective analyses focusing on both wild type and novel mutations in pancreatic cancer and their clinical implications.
Next-Generation Sequencing in Pancreatic Cancer: Revealing Genomic Mutations Beyond KRAS (Abstract #208 [8])
This is a retrospective analysis of patients with pancreatic adenocarcinoma who had next generation sequencing. Next generation sequencing is a newer method of DNA sequencing that enables rapid sequencing of large stretches of DNA base pairs spanning entire genomes. Researchers can now sequence more than five human genomes in a single run, producing data in approximately one week at a relatively cheaper cost of $5,000 per genome (as compared to the much less efficient and expensive capillary electrophoresis based Sanger technology that was used in the human genome project in 2003).
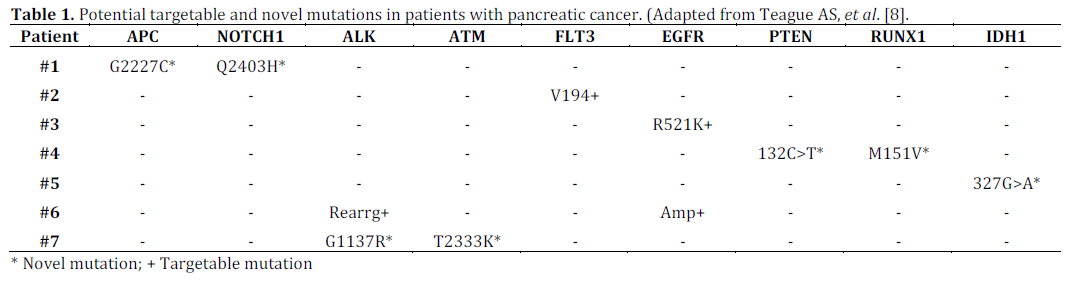
The main genes included in this study were APC, ATM, BRAF, FLT3, NOTCH1, MET, FGFR4, MAPK1, MAP2K2, IDH1, IDH2, RET, JAK2, KRAS, NRAS, PTEN, EGFR, PIK3CA, PDGFRA, RUNX1, KIT, ALK, and TP53. Fluorescence in-situ hybridization (FISH) for EGFR gene amplification and MLL and ALK gene rearrangement were also performed. In the analysis, 36 out of 46 patients (78.3%) had KRAS mutations and 73.9% had TP53 mutations. Out of ten patients with wild type KRAS, 7 had either novel or potential targetable mutations (as shown in Table 1). Among 33 novel mutations not previously described in the Catalogue of Somatic Mutations in Cancer (COSMIC) database (an online database of somatic gene mutations in human cancers; https://cancer.sanger.ac.uk), more than 10% were predicted to have deleterious effect by Protein Variation Effect Analyzer (PROVEAN; https:// provean.jcvi.org) and SIFT (https://sift.jcvi.org) programs (J. Craig Venter Institute, Rockville, MD, USA) that predict whether amino acid substitutions impact biologic function of proteins. Three of twenty-nine patients whose tumors underwent FISH had EGFR gene amplification, one had both EGFR gene amplification and ALK gene rearrangement, and one had MLL gene rearrangement. Thus, next generation sequencing of pancreatic cancer confirmed common mutations like KRAS and TP53 and reaffirmed a vast array of novel mutations that have yet to be studied in detail or targeted for potential drug therapy (Table 1).
Clinicopathological Features of Patients with KRAS Wild-Type Pancreatic Adenocarcinoma (Abstract #192 [9])
Ninety percent of tumors have activating mutations in the KRAS2 oncogene. Transcription of the mutant KRAS oncogene produces an abnormal ras protein that is “locked” in activated form, resulting in activation of proliferative and survival signaling pathways [6]. KRAS2 mutation likely limits the action of epidermal growth factor receptor (EGFR) inhibitor, erlotinib, which is the only agent in combination with gemcitabine that has been shown to have survival benefit in advanced pancreatic cancer [6]. This makes KRAS2 an attractive target for novel therapeutic agents.
This is a retrospective review of 71 patients with pancreatic cancer diagnosed between 2009 and 2012. The KRAS codon 12 and 13 and BRAF codon 600 mutations were assessed in diagnostic tumor tissues using Mutector II Shifted Termination Assay (TrimGen Co., Sparks, MD, USA). KRAS mutations were identified in 50 tumors (70%), while 21 tumors (30%) had KRASwt genotype. Almost all (96%) mutations occurred at codon 12; there were no mutations in codon 13, and none of the KRASwt tumors harbored BRAF mutations. Patients with KRASwt tumors were mostly white (76%), females (57%), did not have diabetes mellitus (80%) and had lower CA 19-9 values (median CA19-9: 392 vs. 3371 U/mL; P=0.042). They were also younger at diagnosis (median age of 60 vs. 66 years; P=0.1) although this was not statistically significant. Head of the pancreas was the primary site of disease in 58% of patients with KRASwt tumors and the body or tail in 53% with KRAS mutant tumors. Although slightly more than 50% of patients presented with distant metastases or locally advanced disease, liver involvement was seen in only 55% of patients with KRASwt tumors compared to 92% in the patients with mutant KRAS tumors (P=0.016). In a univariate analysis, age was an independent predictor of overall survival (HR=1.14; P=0.037) in patients with KRASwt tumors, while level of bilirubin at presentation was a predictor of overall survival (HR=1.86, 95% CI: 0.77-4.53; P<0.01) in patients with KRASwt tumors who had distant metastasis. This study showed that patients with KRASwt and KRAS mutant type had a biologically different disease process.
Discussion
The abstracts reviewed emphasize the genetic heterogeneity of pancreatic cancer with a likely window into how genetic differences translate into biologic variation.
In the first abstract (Abstract #208 [8]), only 46 patients had adequate tissue for analysis and of these only 29 patients underwent FISH and EGFR amplification. No demographic, clinical or pathologic data is available to correlate existence of these mutations to outcomes. Only 10% of these mutations had a deleterious impact when analyzed by PROTEAN and SIFT though the effect of this number is uncertain. Larger clinical settings and more robust clinical data are needed to correlate mutations to biologic effect and clinical outcomes. This is particularly true in the age of new and improved DNA sequencing techniques that give us access to tremendous amounts of genetic information rapidly and more cost-effectively, making it challenging to sort through for clinically relevant information. More studies are needed to obtain details about other potentially targetable mutations seen in pancreatic cancer. Despite the above limitations, these gene targets seem promising. A multimodality approach targeting multiple signaling pathways might also help overcome drug resistance.
In the second abstract (Abstract #192 [9]), biologic and clinical differences are seen with KRAS mutations, comparing the wild type and mutant type. This is the most common mutation seen in pancreatic cancer and the study reaffirms that KRAS2 is a very attractive target for future drugs and possibly decreasing resistance of tumors to erlotinib. To reiterate, this is a small study and results may not be generalized but it effectively brings out clinical effects of KRAS mutations. More studies are needed for clinical correlation with other common and novel mutations to ascertain clinical outcomes, including overall survival and disease free survival. The results of these studies hold potential not only for targeted therapeutics but also for diagnostic and prognostic purposes.
This discussion would be incomplete without mentioning the importance of tissue banks and developing nation-wide consortiums and registries. This collaboration would not only provide us with a true sample to fully elucidate the genetic mutations associated with this complex disease, but we would also be able to gather standardized information to make clinical decisions and truly transform this exciting data from bench to bedside.
The era of personalized medicine in upon us: customizing chemotherapy according to each patient’s characteristic genome. It also has the potential to target therapy to individuals who would be most likely to have a favorable response making it more efficacious and cost effective. This is particularly promising for pancreatic cancer, which currently portends a very poor prognosis. However, there is much to be done in this field and more studies are needed to bring this concept to reality.
Financial support
None
Conflict of interest
The authors hereby state that there is no conflict of interest to disclose during the time of submission of this manuscript. We have no affiliation, financial agreement or other involvement of any author to disclose.
References
- Howlader N, Noone AM, Krapcho M, Garshell J, Neyman N, Altekruse SF, Kosary CL, Yu M, Ruhl J, Tatalovich Z, Cho H, Mariotto A, Lewis DR, Chen HS, Feuer EJ, Cronin KA (eds). SEER Cancer Statistics Review, 1975-2010. https://seer.cancer.gov/csr/1975_2010/
- Mahalingam D, Kelly K, Giles FJ et al. Emerging drugs in the treatment of pancreatic cancer. Expert Opin Emerging Drugs (2009);14(2):311-328.
- Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat Med 2004;10:789-99.
- Jones S, Zhang X, Parsons DW, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 2008;321:1801-6.
- Buchholz M, Gress TM. Molecular changes in pancreatic cancer. Expert Rev Anticancer Ther 2009;9:1487-97.
- Hidalgo M. Pancreatic cancer. New Engl J Med 2010;362:1605-07.
- Iacobuzio-Donahue 2011. Genetic evolution of pancreatic cancer: lessons learnt from the pancreatic cancer genome sequencing project. Gut 2012;G1:1085-94.
- Teague AS, Tan BR, Wang-Gilliam A et al. Next-generation sequencing in pancreatic cancer: Revealing genomic mutations beyond KRAS. J Clin Oncol 32, 2014 (suppl 3; abstr 208)
- Salem ME, Al-hajeili MR, Karmanos BA et al. Clinicopathologic features of patients with KRAS wild-type pancreatic adenocarcinoma. J Clin Oncol 32, 2014 (suppl 3; abstr 192)


