Keywords
Neuroendocrine tumor; Pancreas; Pharmacogenetics
INTRODUCTION
Pancreatic neuroendocrine tumors (panNETs) are uncommon. With an incidence of 1 per 100,000 per year, it represents approximately three percent of primary pancreatic neoplasms [1, 2]. While NETs can occur anywhere in the gastrointestinal tract, those arising from the pancreas typically have a more aggressive course [3]. Most patients present in the fourth to sixth decade of their lives and are usually sporadic; however, NETs may be a part of a hereditary syndrome. These tumors can range from being well differentiated and nonfunctional to being poorly differentiated and highly aggressive. Grading of tumor type is based on cell proliferation rate estimated by Ki-67 positivity and mitotic rate. Symptomatic and progressive panNETs are generally treated with cytotoxic chemotherapy, whereas molecular targeted therapy is used for well differentiated tumors.
What Did We Know Before the 2014 ASCO Annual Meeting?
Although panNETs have been extensively studied, the mechanism behind their pathogenesis remains largely elusive. While most cases are sporadic, the following three hereditary syndromes have a high incidence of these tumors: Multiple Endocrine Neoplasia type 1 (MEN1), von Hippel-Lindau (VHL) and von Recklinghausen’s disease (Neurofibromatosis 1) [4]. Table 1 summarizes the gene loci associated with the syndromes and the tumor frequency and character [4].

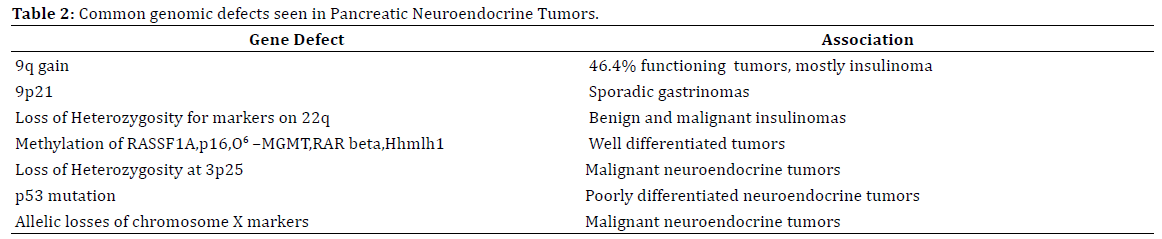
Germline MEN 1 mutation followed by loss of a wild type allele is an inciting event in 33% of nonfunctional panNETs, 10-27% of insulinomas and 39-45% of gastrinomas [4, 5]. Multiple other aberrations in the genome have been noted in various pancreatic neuroendocrine tumors. Table 2 summarizes these mutations and the tumor types.
Since these tumors can have variable manifestations based on anatomic location, degree of differentiation and functionality, different treatment approaches can be undertaken. For well differentiated tumors that are not surgically resectable but asymptomatic, molecular targeted therapy or suppression of secretory function (if they are functional) are proposed approaches. Systemic cytotoxic therapy is suggested for large well differentiated tumors that may cause visceral symptoms. Poorly differentiated panNETs are treated with platinum based chemotherapy, similar to small cell cancer of the lung.
Somatostatin analogues have been useful in treating VIPoma, Glucagonomas and even Somatostatinomas, but their utility is limited in the management of Gastrinomas and Insulinomas (the most common secretory panNETs). Most Gastrinomas respond to Proton Pump Inhibitors and use of everolimus has shown promise in Insulinomas by counter-acting the hypoglycemic effect of these tumors.
Molecular targeted therapies have been used more recently, with development of agents active against pathways that have been found to be up-regulated in these tumors [6]. VEGF and mTOR inhibitors have been investigated and have found use in panNETs. Sunitinib, a multi-targeted tyrosine kinase inhibitor, has been approved by the FDA in the USA for the treatment of progressive, welldifferentiated panNETs in patients with unresectable, locally advanced, or metastatic disease. Two other orally active tyrosine kinase inhibitors, sorafenib and pazopanib, have demonstrated modest activity in panNETs in phase II studies [7].
Everolimus has been approved by the FDA based on results from the RADIANT-3 trial of 410 patients with advanced progressing panNETs. Everolimus was associated with a significant prolongation in median PFS (11.0 versus 4.6 months, hazard ratio [HR] for progression 0.35, 95% CI 0.27 to 0.45) when compared to placebo [8]. It is currently used for the treatment of progressive NETs of pancreatic origin in patients with unresectable, locally advanced, or metastatic disease. As hyperglycemia is a common side effect, the drug has also found to be useful in treatment of Insulinomas with refractory hypoglycemia.
Patients with symptoms secondary to bulky panNET or have rapidly progressing metastatic disease are recommended to receive systemic cytotoxic chemotherapy. Streptozocin and Dacarbazine have historically been used but have been less favored lately in comparison to temozolomide due to toxicity. Temozolomide is a less toxic orally active analog of dacarbazine with activity in panNETs. It has been used in combination with thalidomide, bevacizumab, everolimusor capecitabine. Table 3 summarizes the agents being used for panNETs.

Methylguanine DNA methyltransferase (MGMT) is an enzyme that is responsible for DNA repair induced by alkylating agent chemotherapy (Figure 1) [9]. Low expression has been correlated with better response to Temozolomide. A decreased level is seen more commonly in panNETs than other intestinal NETs and is cited as a probable reason for better responses in the former group.

Figure 1. Cellular processing and repair of alkylating agent induced O6meG lesions in DNA by MGMT. SN1-alkylating agents induce the formation of O6-methylguanine (O6meG), which is directly repaired by O6-methylguanine-DNA methyltransferase (MGMT). If the O6meG lesion is not repaired, DNA
replication by either conventional or translesion DNA synthesis (TLS) polymerases can lead to the formation of O6meG: Tmispairs. Recognition of O6meG:
T by the MUTSα protein complex initiates the mismatch repair (MMR) pathway, leading to futile cycles of DNA resection and processing that can cause
replication fork collapse and double-strand DNA break (DSB) formation. The cytotoxic DSB can be repaired by the homologous recombination (HR) or nonhomologous
end-joining (NHEJ) pathways resulting in cell survival but with mutation or sister chromatid exchange (SCE). If the DSB is not repaired, it can
trigger programmed cell death by apoptosis. Alternatively, MMR-dependent recognition of O6meG:T can directly signal for programmed cell death through
the ataxia-telangiectasia mutated and Rad3-related (ATR)-ATR-interacting protein (ATRIP) signalling pathway. (With permission from Nature Reviews,
License Number: 3405920932825)
What Did We Learn at the 2014 ASCO Annual Meeting?
We summarize here two abstracts that were presented at the ASCO Annual Meeting 2014 in Chicago on Pharmacogenetics in neuroendocrine tumor of the pancreas.
Profiling of 1,350 Neuroendocrine Tumors for Identification of Multiple Potential Drug Targets. (Abstract #4113)
Astsaturov et al. evaluated the utility of genomic profiling of 1350 cases of pancreatic neuroendocrine tumor in attempt to identify new potential drug targets[10]. Using multiple modalities for genetic profiling, including gene sequencing, gene copy number and protein expression, drug-related alterations were identified in a majority of samples. Several case examples of durable response were reported in a patient with a Kit mutation, responsive to Imatinib, and a patient with low MGMT and Low TS, treated with streptozocin or temozolomide plus fluoropyrimidine chemotherapy. Other markers assessed included RRM1, BRAF (both the V600E and G596R mutations), CTNNB1, EGFR, FGFR2, GNAS, HRAS, PIK3CA, RB1, VHL, KRAS, NRAS, APC, and MET. The authors conclude that while individual genetic mutations occur with low frequency, most patients exhibit at least one mutation that could potentially be used as a target for therapy.
MGMT Immunohistochemistry (IHC) and Exclusion of Pancreatic NET (PanNET) Patients from Treatment with Temozolomide Based Therapy (Abstract #e15169)
This is a retrospective analysis of tissue from 13 patients with panNET that responded to treatment with Temozolomide and Capecitabine [11]. Reidy et al. reportedan institutional review of panNET samples from patients treated with Capecitabine and Temozolomide (C/T). The aim of the study was to determine if tumor 06-methylguanine-methyltransferase (MGMT) expression correlated with effect of C/T chemotherapy. Thirteen patients who had MGMT positive tumors were evaluable, of whom 4 (31%) demonstrated a clinical response to chemotherapy. Nine patients were MGMT negative, and all demonstrated tumor response. Burden of disease, tumor grade, and number of systemic treatments prior to resection did not correlate with tumor response. The authors conclude that while absence of MGMT correlates with a response to chemotherapy with C/T, 30% of those that were MGMT positive also demonstrated a response, indicating a need to identify more sensitive and prognostic predictors.
Discussion
The abstracts reviewed here underscore the importance of clinically relevant information one can potentially obtain from molecular analysis by not only providing potential targets for novel therapeutics but also in selecting patients more appropriately for systemic treatment.
The first abstract (#4113) identifies gene products that can help predict response to systemic treatment. Although MGMT expression was low or absent in over 61 % of panNET patients, similar organ specific stratification was not done for low/absent RRM1 and thymidine synthase expression. Multiple genetic mutations cited as potential targets may be useful but their frequencies were low. As with other solid tumors, mutation targeted therapy does not always result in a desirable response, as most of these pathways tend to inter-digitate and tumor tends to escape the blockade. Additionally, cost of biomarker profiling should be kept in mind and how this would affect the health system still needs to be evaluated.
Although small in numbers, the second abstract (#e15169) highlights responders to Temozolomide/Capecitabine that do not have absent MGMT expression. Therefore lack of MGMT expression by IHC cannot be universally used as a marker predictive of response to treatment. It must however be mentioned that the abstract did not provide the strength of positivity in the 4 responders; this may be of value, given that patients with low (+1 by IHC) level expression have been found to respond.
Given the rare occurrence of this tumor type and potentially aggressive behavior, better therapies are certainly needed. Targeted therapy has been touted as the wave of the future; however success rates in solid tumors have overall been modest. Certainly, better prediction of treatment response at this time appears to show promise; however, complexities of genetic alterations and cellular pathways need to be elucidated more to help identify targets that can be utilized clinically in a meaningful way.
Conflict of Interest
The authors have no potential conflicts of interest.
References
- Klöppel G, Perren A, Heitz PU. The gastroenteropancreatic neuroendocrine cell system and its tumors: the WHO classification. Ann N Y AcadSci 2004; 1014: 13-27.
- Fesinmeyer MD, Austin MA, Li CI, De Roos AJ, Bowen DJ. Differences in survival by histologic type of pancreatic cancer. Cancer Epidemiol Biomarkers Prev 2005; 14: 1766-1773. [PMID:16030115]
- Panzuto F, Nasoni S, Falconi M, Corleto VD, Capurso G, Cassetta S, Di Fonzo M, et al. Prognostic fa [ctors and survival in endocrine tumor patients: comparison between gastrointestinal and pancreatic localization. EndocrRelat Cancer 2005; 12: 1083-1092. [PMID:16322345]
- Lubensky IA, Zhuang Z. Molecular genetics events in gastrointestinal and pancreatic neuroendocrine tumors. EndocrPathol 2007; 18: 156- 162.
- Zhuang Z, Vortmeyer AO, Pack S, Huang S, Pham TA, Wang C, Park WS, et al. Somatic mutations of the MEN1 tumor suppressor gene in sporadic gastrinomas and insulinomas. Cancer Res 1997; 57: 4682-4686. [PMID:9354421]
- Oberg K, Casanovas O, Castaño JP, Chung D, DelleFave G, Denèfle P, Harris P, et al. Molecular pathogenesis of neuroendocrine tumors: implications for current and future therapeutic approaches. Clin Cancer Res 2013; 19: 2842-2849. [PMID:23459719]
- Raymond E, Hobday T, Castellano D, Reidy-Lagunes D, García- Carbonero R, Carrato A. Therapy innovations: tyrosine kinase inhibitors for the treatment of pancreatic neuroendocrine tumors. Cancer Metastasis Rev 2011; 30 Suppl 1: 19-26. [PMID:21308478]
- Yao JC, Phan AT, Jehl V et al. Everolimus in advanced pancreatic neuroendocrine tumors: the clinical experience. Cancer Res 2013; 73: 1449-1453.
- Fu D, Calvo JA, Samson LD. Balancing repair and tolerance of DNA damage caused by alkylating agents. Nat Rev Cancer 2012; 12: 104-120.
- Igor A. Astsaturov SJC, Paul F. Engstrom, Sherri Z. Millis. Profiling of 1,350 neuroendocrine tumors for identification of multiple potential drug targets. J ClinOncol 2014; 32:5s, 2014 (suppl; abstr 4113).
- Reidy DLOB, Kriplani A, Abou-Alfa GK, Jarnagin WR, Allen PJ, Saltz L, Klimstra DS, et al. MGMT immunohistochemistry (IHC) and exclusion of pancreatic NET (PanNET) patients from treatment with temozolomide-based therapy. J Clin Oncol 2014; 32, 2014 (suppl; abstr e15169).

