Keywords
alpha-Fetoproteins; Beckwith-Wiedemann Syndrome; Cisplatin; Doxorubicin; Hepatoblastoma; Infant; Pancreatic Neoplasms
Abbreviations
LOH: loss of heterozygosity; PB: pancreatoblastoma
Summary
Pancreatoblastoma (PB), or infantile pancreatic carcinoma, is an extremely rare pancreatic tumor in childhood, comprising 0.5% of pancreatic non-endocrine tumors. Although PB mainly presents during childhood but can also occur in adults. PB tend to be less aggressive in infants and children compared to adults. Children with PB usually present late with upper abdominal pain and many have a palpable mass in the epigastrium. Mechanical obstruction of the upper duodenum and gastric outlet by tumor in the head of the pancreas may be associated with vomiting, jaundice and gastrointestinal bleeding. Histologically, PB is characterized with distinct acinar and squamoid cell differentiation. PB has been associated with alterations in the Wnt signaling pathway and chromosome 11p loss of heterozygosity (LOH), Beckwith-Wiedemann syndrome and familial adenomatous polyposis. The majority of these tumors arise in the head of the pancreas. Alpha-fetoprotein may be elevated in up to 68% of patients with PB. Ultrasound and CT scan may be useful but preoperative diagnosis is often quite difficult. The treatment of choice is complete resection, that may often be curative. The role of adjuvant chemotherapy or radiotherapy is still under discussion due to small number of patients treated as yet. Chemotherapy regimens consisting of cyclophosphamide, etoposide, doxorubicin, and cisplatin have been used in neoadjuvant setting with anecdotal benefit. Prognosis of this rare tumor is good, when resected completely. Prognosis is poorer, when there is metastasis or when it is inoperable. On the whole, PB is regarded to be a curable tumor; hence the clinical diagnosis should be made early. Awareness of this rare tumor of pancreas is essential for early detection and proper management. The author review the clinical presentation, etiology, diagnosis, treatment and prognosis of PB in this presentation [1].

[1]

Pancreatoblastoma (PB), is an extremely rare pancreatic tumor of childhood, but can occur in adults. PB often exhibits elevated plasma levels of alpha-fetoprotein. PB, though not common, is said to be less aggressive in infants and children compared to adults [2, 3, 4].

PB is an extremely rare pancreatic tumor of childhood. The term PB was coined by Horie et al. in 1977 to describe tumors previously known as “infantile carcinoma of the pancreas” [5]. PB has several similarities to hepatoblastoma, including association with the Beckwith-Wiedemann syndrome and elevated plasma levels of alpha-fetoprotein (AFP) [6].

Pancreatic tumors are rare in children, and PB comprises only 0.5% of pancreatic non-endocrine tumors occurring in children. This tumor is more common in Asians than in the white population. They have also been diagnosed in-utero and in adults, with the oldest patient being 68-year-old. [2, 7, 8, 9]

The presenting complaints are varied (for instance we observed a patient who had no prior complaints and was identified following investigations after a motor vehicle accident). Children with PB usually present late with upper abdominal pain and many have a palpable mass in the epigastrium [1]. Mechanical obstruction of the upper duodenum and gastric outlet by tumor in the head of the pancreas can complicate with obstruction and gastrointestinal bleeding. Poor nutritional intake and the resultant weight loss may also be found.

Largest size reported in the literature reviews has been around 15 cm. Majority of the tumors are encapsulated, while the rest are partially encapsulated. Encapsulated tumors have a better prognosis. [1, 3]
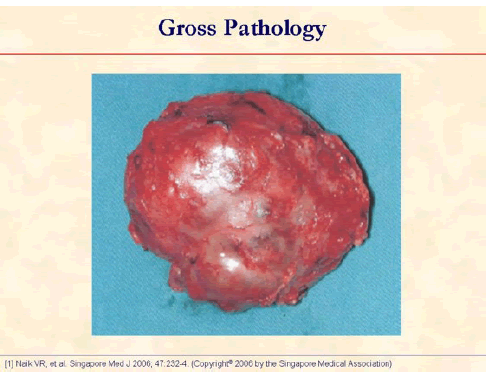
The specimen photograph shows an encapsulated tumor with a nodular surface. The capsule is complete [1].

Molecular investigation has disclosed a mosaic paternal 11p15 uniparental disomy in the tumor cells of PB. Recently genetic alterations also have been characterized and the commonest change is allelic loss of 11p. Familial adenomatous polyposis and Beckwith-Wiedemann syndrome have also een associated with PB. [6, 10, 11, 12, 13]

It is said to be that hamartomatous or dysembryogenic development of ductal cells of ventral portion of primordial pancreas lead to the development of PB. PB contains pluripotent cells capable of differentiating along the pathway of all three pancreatic cell types: acinar, endocrine and ductal. [14]

PB can exhibit acinar, endocrine and ductal differentiation. Histopathological features that are readily seen include hemorrhage, capsule formation and necrosis. [14, 15, 16]
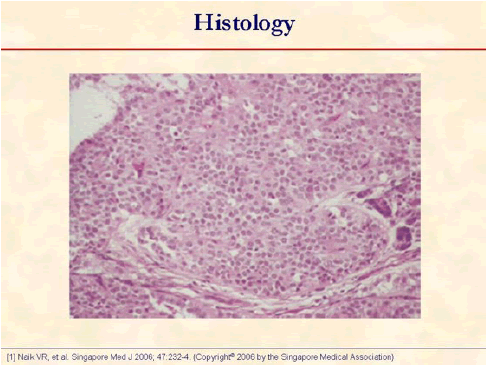
Photomicrograph shows a cellular tumor with uniform epithelial cells arranged in nests and acini (hematoxylin and eosin, x250) [1].

Immunohistochemistry is usually strongly positive for alpha-1-antitrypsin and glucose-6-phosphatase, in addition acid phosphatase, esterase and enteroprotease activity may be demonstrated using histochemistry. Stains for chromogranin, synaptophysin and neuron-specific enolase are often positive. Trypsin and chymotrypsin are usually found in acinar regions but positivity for specific peptide hormones is rare. Immunohistochemistry for AFP may be positive within solid regions of the epithelial component. Electron microscopy reveals multiple cytoplasmic neurosecretory zymogen granules. [15, 16]
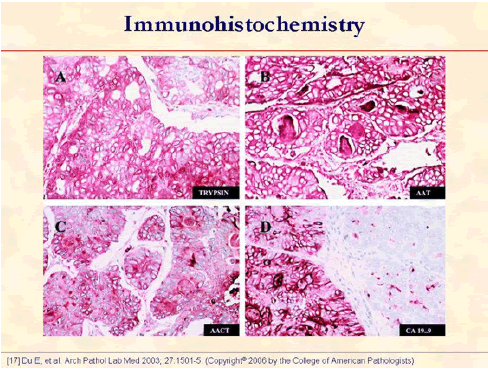
The tumor cells show strong positive immune reaction for trypsin (A), alpha1-antitrypsin (B), alpha1-antichymotrypsin (C), and CA 19-9 (D). Note that the luminal contents are positive for alpha1-antitrypsin (B), alpha1-antichymotrypsin (C), and CA 19-9 (D) (original magnifications x400) [17].

Biochemistry and radiology are the main diagnostic tools in PB.

AFP is the most commonly used tumor marker in PB. Other tumor markers do not show any significant correlation. [2, 4, 18, 19]

Elevated serum AFP levels have been reported in up 68% of cases. AFP level comes down once the tumor is resected. [4, 18]

Radiological staging include a CT scan of abdomen, pelvis and chest. Bone scan or a brain MRI may be performed if clinically indicated. [2, 7, 8, 14]
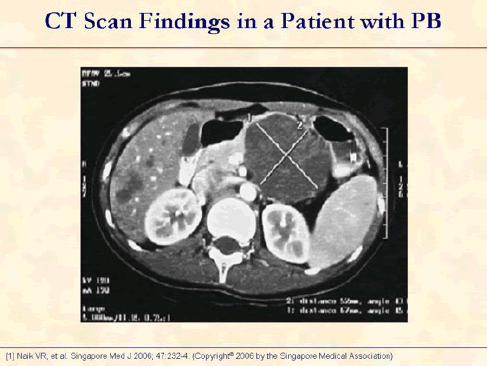
Enhanced axial CT image shows a large well-defined cystic tumor in the tail of pancreas and multiple small nodules in the liver [1].

The most common cystic pancreatic tumors in children are microcystic adenomas and cystadenocarcinomas.
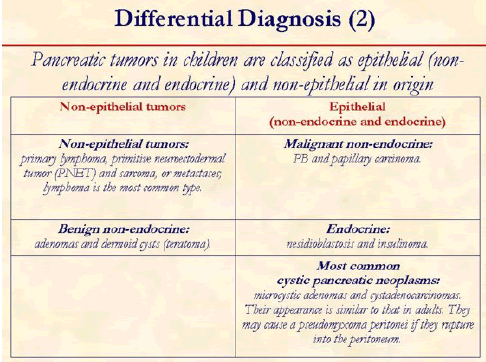
Differentiation of PB from other tumors is extremely important as prognosis of this rare tumor is good, when resected completely.

Clinically PB can be distinguished from the following neuroendocrine tumors due to their different spectrum of symptoms. Insulinoma: hypoglycemia, behavior change, weight gain and/or morning seizures; Gastronoma: severe gastrointestinal ulceration and diarrhea; VIPoma: watery diarrhea, hyperkalemic and achlorhydia; Glucagonomas: migratory necrolytic dermatitis, weight loss, stomatitis, anemia and hyperglycemia; Somatostatin-omas: diarrhea and may develop diabetes mellitus.


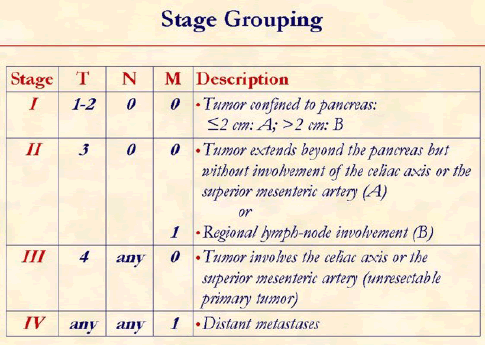
The tumor, node, metastasis (TNM) classification of the American Joint Committee on Cancer is usually used to determine the tumor staging [20].


PB is less aggressive in infants and children compared to adults. Prognosis of PB is good, when resected completely. Prognosis is poorer, when there is metastasis or when it is inoperable. [2, 7, 21]

PB has an indolent course and is amenable for various modes of treatment but surgery is the most optimal treatment.


Complete resection of the tumor offers the best prognosis. However, in the presence of metastatic disease, it is of limited value. [1, 2, 3, 7, 8, 9, 22]





In these situations where there are suspected or documented metastatic lesions, empirical chemotherapy regimens that include cisplatin and doxorubicin have been used. A higher rate of metachronous metastasis has been reported in patients undergoing chemotherapy. [1, 2, 3, 7, 8, 9, 23, 24, 25, 26]

When the tumor is unresectable and the patient is non-responsive to chemotherapy, radiotherapy is given. Shrinkage of PB has been reported after treatment with radiotherapy [27].

[8, 16, 17]


Acknowledgements
We thank Dr. Naik [1] and Dr. Shabaik [17] for allowing us to reproduce figures from their publications. The figures have been reprinted with permission from: Singapore Medical Journal (SMJ), Copyright© 2006 by the Singapore Medical Association [1]; and Archives of Pathology and Laboratory Medicine, Copyright© 2006 by the College of American Pathologists [17]
References
- Naik VR, Jaafar H, Leow VM, Bhavaraju VM. Pancreatoblastoma: a rare tumour accidentally found. Singapore Med J 2006; 47:232-4. [PMID 16518559]
- Brennan B. Pancreatoblastoma. Orphanet Encyclopedia. August 2004. (Accessed September 7th, 2006).
- Kohda E, Iseki M, Ikawa H, Endoh M, Yokoyama J, Mukai M, et al. Pancreatoblastoma. Three original cases and review of the literature. Acta Radiol 2000; 41:334-7. [PMID 10937753]
- Morohoshi T, Sagawa F, Mitsuya T. Pancreatoblastoma with marked elevation of serum alpha-fetoprotein. An autopsy case report with immunocytochemical study. Virchows Arch A Pathol Anat Histopathol 1990; 416:265-70. [PMID 1689089]
- Horie A, Yano Y, Kotoo Y, Miwa A. Morphogenesis of pancreatoblastoma, infantile carcinoma of the pancreas: report of two cases. Cancer 1977; 39:247-54. [PMID 188539]
- Koh TH, Cooper JE, Newman CL, Walker TM, Kiely EM, Hoffmann EB. Pancreatoblastoma in a neonate with Wiedemann-Beckwith syndrome. Eur J Pediatr 1986; 145:435-8. [PMID 3792392]
- Dhebri AR, Connor S, Campbell F, Ghaneh P, Sutton R, Neoptolemos JP. Diagnosis, treatment and outcome of pancreatoblastoma. Pancreatology 2004; 4:441-53. [PMID 15256806]
- Levey JM, Banner BF. Adult pancreatoblastoma in an elderly patient: A case report and review of literature. Am J Gastroenterol 1996; 91:1841-4. [PMID 8792711]
- Defachelles AS, Martin De Lassalle E, Boutard P, Nelken B, Schneider P, Patte C. Pancreatoblastoma in childhood: clinical course and therapeutic management of seven patients. Med Pediatr Oncol 2001; 37:47-52. [PMID 11466723]
- Kerr NJ, Chun YH, Yun K, Heathcott RW, Reeve AE, Sullivan MJ. Pancreatoblastoma is associated with chromosome 11p loss of heterozygosity and IGF2 overexpression. Med Pediatr Oncol 2002; 39:52-4. [PMID 12116082]
- Drut R, Jones MC. Congenital pancreatoblastoma in Beckwith-Wiedemann syndrome: an emerging association. Pediatr Pathol 1988; 8:331-9. [PMID 2845376]
- Potts SR, Brown S, O'Hara MD. Pancreoblastoma in a neonate associated with Beckwith-Wiedemann syndrome. Z Kinderchir 1986; 41:56-7. [PMID 3962517]
- Abraham SC, Wu TT, Klimstra DS, Finn LS, Lee JH, Yeo CJ, et al. Distinctive molecular genetic alterations in sporadic and familial adenomatous polyposis-associated pancreatoblastomas : frequent alterations in the APC/beta-catenin pathway and chromosome 11p. Am J Pathol 2001; 159:1619-27. [PMID 11696422]
- Klimstra DS, Wenig BM, Adair CF, Heffess CS. Pancreatoblastoma. A clinicopathologic study and review of the literature. Am J Surg Pathol 1995; 19:1371-89. [PMID 7503360]
- Silverman JF, Holbrook CT, Pories WJ, Kodroff MB, Joshi VV. Fine needle aspiration cytology of pancreatoblastoma with immunocytochemical and ultrastructural studies. Acta Cytol 1990; 34:632-40. [PMID 2220242]
- Palosaari D, Clayton F, Seaman J. Pancreatoblastoma in an adult. Arch Pathol Lab Med 1986; 110:650-2. [PMID 3013120]
- Du E, Katz M, Weidner N, Yoder S, Moossa AR, Shabaik A. Ampullary pancreatoblastoma in an elderly patient: a case report and review of the literature. Arch Pathol Lab Med 2003; 127:1501-5. [PMID 14567752]
- Bergstraesser E, Ohnacker H, Stamm B, Angst R, Imbach P, Gnehm HE. Pancreatoblastoma in childhood: the role of alpha-fetoprotein. Med Pediatr Oncol 1998; 30:126-7. [PMID 9403024]
- Rajpal S, Warren RS, Alexander M, Yeh BM, Grenert JP, Hintzen S, et al. Pancreatoblastoma in an adult: case report and review of the literature. J Gastrointest Surg 2006; 10:829-36. [PMID 16769539]
- Exocrine pancreas. In: Greene FL, Page DL, Fleming ID, Fritz A, Balch CM, Haller DG, Morrow M, eds. American Joint Committee on Cancer: AJCC Cancer Staging Manual. 6th ed, New York, NY, USA: Springer, 2002:157-164. [ISBN 0-387-95271-3]
- Benoist S, Penna C, Julie C, Malafosse R, Rougier P, Nordlinger B. Prolonged survival after resection of pancreatoblastoma and synchronous liver metastases in an adult. Hepatogastroenterology 2001; 48:1340-2. [PMID 11677959]
- Caracciolo G, Vicedomini D, Di Blasi A, Indolfi P, Casale F, De Dominicis G, et al. Adenocarcinoma of the pancreas in childhood (pancreatoblastoma): report of a case with good response to chemotherapy. Tumori 1995; 81:391-4. [PMID 8804461]
- Vannier JP, Flamant F, Hemet J, Caillaud JM, Gruner M, Bachy B, et al. Pancreatoblastoma: response to chemotherapy. Med Pediatr Oncol 1991; 19:187-91. [PMID 1708850]
- Inomata Y, Nishizawa T, Takasan H, Hayakawa T, Tanaka K. Pancreatoblastoma resected by delayed primary operation after effective chemotherapy. J Pediatr Surg 1992; 27:1570-2. [PMID 1469578]
- Ogawa B, Okinaga K, Obana K, Nakamura K, Hattori T, Ito T, et al. Pancreatoblastoma treated by delayed operation after effective chemotherapy. J Pediatr Surg 2000; 35:1663-5. [PMID 11083448]
- Perilongo G, Brown J, Shafford E, Brock P, De Camargo B, Keeling JW, et al. Hepatoblastoma presenting with lung metastases: treatment results of the first cooperative, prospective study of the International Society of Paediatric Oncology on childhood liver tumors. Cancer 2000; 89:1845-53. [PMID 11042582]
- Griffin BR, Wisbeck WM, Schaller RT, Benjamin DR. Radiotherapy for locally recurrent infantile pancreatic carcinoma (pancreatoblastoma). Cancer 1987; 60:1734-6. [PMID 3115557]

