Keywords
Adenocarcinoma; Ampulla of Vater; Mixed Tumor, Malignant; Neurofibromatosis 1
INTRODUCTION
Mixed endocrine tumors are rare, especially those arising in the ampulla of Vater. Ampullary somatostatinomas are frequently associated with neurofibromatosis type 1. We report the first case of a mixed endocrine somatostatinoma associated with neurofibromatosis type 1. These tumors raise several difficulties. The first one is to diagnose this rare tumor, composed of both endocrine and glandular components. The second difficulty is the management of this double neoplasm which is often malignant and frequently requires surgical resection with neoadjuvant chemotherapy. Because of their rarity, ampullary mixed endocrine tumors may be misdiagnosed and receive incorrect therapeutic treatment.
CASE REPORT
A 49-year-old woman with neurofibromatosis type 1 presented with atypical abdominal pain. Endoscopic examination revealed a duodenal villous tumor involving the ampulla of Vater. A CT scan showed a 2 cm ampullary tumor with stenoses of both the distal common bile duct and the main pancreatic duct (Figure 1a). Neither lymph node nor distant metastases were found. Endoscopic ultrasound identified an ampullary tumor with some papillary projections in the juxtaampullary main pancreatic duct and a slight upstream biliary dilatation. Analysis of the bioptic specimens revealed a high grade tubulo-villous adenoma without invasive adenocarcinoma.
Figure 1. a. CT-scan slice at the level of the ampulla, obtained
during the arterial phase after an intravenous injection of contrast
medium, identified a large ampullary and peri-ampullary tumor
invading the duodenum (large black arrow). This tumor is
heterogeneous with a distinct hyperdense nodule (white arrow). The
common bile duct (*) is dilated. b. The large exophytic duodenal
tumor is situated in and around the ampulla of Vater, measuring
4.5x5.0 centimeters. At the center of this tumor, in close contact with
the ampulla of Vater, a well delineated red-brown nodule measuring
12 mm and corresponding to the endocrine component is visible.
A pancreaticoduodenectomy was performed, showing an exophytic duodenal tumor invading the ampulla of Vater, measuring 4.5x5.0 centimeters. At the center of this tumor, a well delineated red-brown nodule measuring 12 mm was observed (Figure 1b). Microscopic examination identified two distinct tumor cell populations. The first one corresponded to an exocrine high grade tubulo-villous adenoma largely extending onto the external part of the ampulla and to the duodenal mucosa (Figure 2a). An area of well- differentiated adenocarcinoma was identified inside the adenoma, measuring 6 mm, infiltrating the duodenal submucosa but sparing the muscularis propria (Figure 2b). The second tumor cell population was of endocrine type. It formed a well-delineated central nodule of 1 cm, sharply demarcated from the exocrine component (Figure 2c). It was composed of cords or acini of well-differentiated endocrine cells combined with psammoma bodies (Figure 2d). Endocrine cells contained regular nuclei, and granular and eosinophilic cytoplasm. Mitotic activity was low (less than 2 mitoses per 10 high-power fields).
Figure 2. a. The high grade tubulo-villous adenoma extending to the duodenal mucosa (H&E staining, x200). b. An area of well differentiated
adenocarcinoma infiltrating the duodenal submucosa (H&E, x400). c. The endocrine component (small arrows) is located under the exocrine high
grade adenoma (large arrow) (H&E, x100). d. The endocrine component is well differentiated and contains psammoma bodies (arrows) (H&E,
x400). e. Endocrine tumor cells abundantly and diffusely expressing somatostatin (immunostaining with somatostatin antibody, counterstained with
hematoxylin; x 200). f. Metastatic lymph nodes are present, either from the exocrine (left) or the endocrine (right) component (H&E, x100).
Immunohistochemical analysis demonstrated positive and diffuse staining of the endocrine component with antibodies directed against the neuroendocrine markers synaptophysin and CD56 and with anti-somatostatin antibody (Figure 2e). Chromogranin A was negative. The proliferation index, evaluated using MIB-1 antibody according to the TNM classification [1] was calculated at 2%. The main pancreatic duct was invaded by the adenomatous high grade component, but the secondary ducts and biliary duct did not display any dysplasia or malignant features. The infiltrating tumors did not extend into the pancreatic parenchyma. Lymph node dissection revealed 6 metastatic nodes: 3 were metastatic from the adenocarcinoma component and 3 from the endocrine carcinoma (Figure 2f).
Due to lymph node metastases from the adenocarcinoma component, chemotherapy with 5- fluorouracil and oxaliplatine was given to the patient; oxaliplatine was stopped after two courses because of liver cytolysis and abdominal ascites without tumor cells. The four following courses were administrated without complications. The patient is alive with no signs of recurrence 3 years after the surgery.
DISCUSSION
Mixed endocrine tumors are tumors composed of at least two distinct tumor populations, one of which is endocrine. Three main types of endocrine mixed tumors are recognized [2, 3]. Collision tumors are formed by two independent tumor populations. Composite tumors are made up of two tumor populations deriving from a common precursor.
Amphicrine tumors are made up of a unique cell population presenting a double differentiation. Composite tumors are the most frequent types of mixed endocrine tumors [2, 3]. The recent WHO classification recommends restricting the term “composite endocrine tumor” to those containing at least 30% of obviously tumoral endocrine cells; some authors recommend using higher thresholds (50%) in order to avoid overdiagnosis [4, 5].The endocrine component is usually well differentiated, easily recognized on standard histology and confirmed with immunohistochemistry by detection of specific endocrine markers (chromogranin A and synaptophysin). When the endocrine component is poorly differentiated, it is more difficult to identify without specific immunohistochemistry.
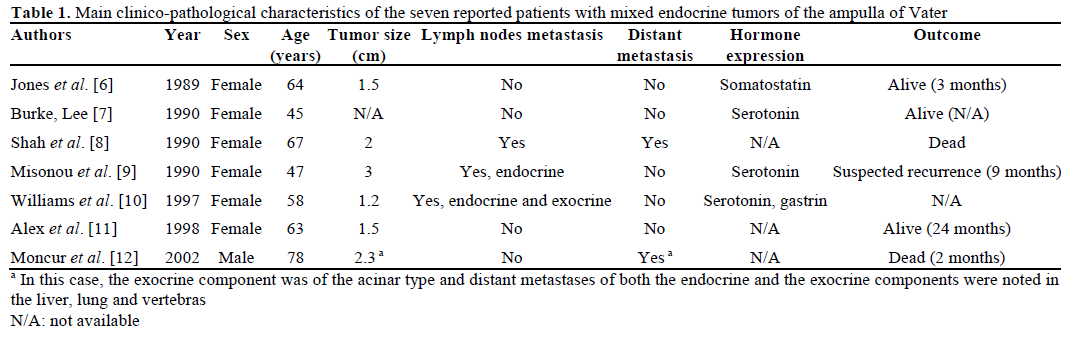
Mixed endocrine tumors have rarely been described in the ampulla of Vater. We reviewed seven cases in the English literature [6, 7, 8, 9, 10, 11, 12] (Table 1).These tumors affected women rather than men (six women versus one man). The median age was 63 years (range: 45-78 years). Tumor size varied from 0.6 to 3 cm. Two patients had lymph node metastasis, including one having both endocrine and exocrine infiltration in the same lymph node; the other patient presented an endocrine lymph node metastasis. Two other patients had widespread metastases (liver, lung and vertebras with both endocrine and glandular components) and died from their disease. Among the endocrine contingents, 3 were well differentiated and 3 were poorly differentiated (differentiation was not noted in one patient). Only one patient had an endocrine component expressing somatostatin, without clinical functional syndrome and without neurofibromatosis [6]. The exocrine component formed glands or tubules with goblet cells and was well-, moderately- and poorly-differentiated in 2, 3 and 2 patients, respectively [12]. In 5 out of 7 cases, the two components were intermixed, in line with the diagnosis of mixed tumors of a composite type. All seven tumors spared the pancreas.
Our case could be considered a collision tumor with two well-differentiated and well-delineated components. In addition, the adenocarcinoma component was associated with a large duodenal highgrade adenoma. The endocrine component contained psammoma bodies and calcifications, a figure which favors the diagnosis of somatostatinoma, the second most frequent duodenal endocrine tumor [13]. The diagnosis was confirmed by the strong and diffuse positivity of somatostatin at immunohistochemistry; somatostatinomas rarely express chromogranin A, as was observed in our case [14]. Many “somatostatinproducing” tumors are not associated with a functional somatostatinoma syndrome and should not be reported as “somatostatinomas” [4, 13, 14, 15]. In our observation, we did not find symptoms due to excessive hormone production and the somatostatinoma was diagnosed incidentally at postsurgical histological examination. However, in some cases, the so-called somatostatinoma syndrome may be present but difficult to recognize as a functional tumor before resection because the clinical syndrome, which associates recent diabetes mellitus and steatorrhea, may be difficult to diagnose. Among the previously reported ampullary somatostatinomas, we did not find any mixed tumors and, to our knowledge, our case is the first reported mixed somatostatinoma associated with a neurofibromatosis type 1 [16, 17, 18, 19, 20]. Ampullary somatostatinomas are classically associated with neurofibromatosis type 1 [14, 18, 20, 21]. Neurofibromatosis type 1 is an autosomallyinherited disease, with an incidence of about 1 per 3,000 births, equally involving males and females. The disease is secondary to mutations in neurofibromatosis type 1 tumor suppressor gene (NF1), encoding the cytoplasmic protein neurofibromin. In neurofibromatosis type 1, some malignancies occur at an increased incidence compared to the general population and an association is recognized with gliomas, ependymomas, malignant peripheral nerve sheath tumors, pheochromocytomas, myeloid leukemia, nephroblastomas, gastrointestinal stromal tumors and endocrine tumors [22]. Digestive endocrine tumors occur in about 1% of neurofibromatosis type 1 patients. They typically arise in the ampullary region of the duodenum [23]. Their exact pathogenic mechanism of development in neurofibromatosis type 1 is not fully understood. In gastrointestinal stromal tumors associated with neurofibromatosis type 1, the loss of heterozygosity of NF1 has been evidenced [24, 25]. Interestingly, there are 6 reported cases of a combination of gastrointestinal stromal tumors and somatostatinomas in patients with neurofibromatosis type 1, suggesting the existence of a common pathway in the development of both diseases [26, 27]. There have been only 2 reports of ampullary adenocarcinomas in neurofibromatosis type 1 [28, 29]. Prognosis may be difficult in mixed tumors. Adenocarcinomas of the ampulla with an intestinal phenotype have a significantly better long-term prognosis than adenocarcinomas located in the head of the pancreas [10]. However, ampullary endocrine tumors have a poorer prognosis than small bowel endocrine tumors and tumor size is a prognostic factor; when greater than 2 cm, the risk of metastasis increases significantly [10, 16, 21, 30].
Our patient had an aggressive tumor with 6 metastatic lymph nodes (of both exocrine and endocrine types). The conventional treatment for both ampullary endocrine and exocrine tumors is surgery; local excision may be indicated for small non-infiltrative lesions. A more radical treatment, i.e. pancreaticoduodenectomy, is appropriate for larger tumors with good long-term survival especially for patients with endocrine peripancreatic metastatic disease [18, 19, 20]. Our patient received adjuvant chemotherapy for the exocrine tumor since this component was considered to be the most aggressive one and since adjuvant chemotherapy is not recommended for resected endocrine tumors
CONCLUSION
We report the first ampullary mixed endocrine tumor with somatostatin expression in a neurofibromatosis type 1 patient. Somatostatin expression and the presence of psammoma bodies in ampullary endocrine tumors must alert the pathologist to the possible association with neurofibromatosis type 1 disease. The management of patients with mixed endocrine tumors is challenging but an early diagnosis allows curative surgical treatment.
Conflict of interest
The authors have no potential conflicts of interest
References
- Rindi G, Klöppel G, Alhman H, Caplin M, Couvelard A, de Herder WW, et al. TNM staging of foregut (neuro)endocrine tumors: a consensus proposal including a grading system. Virchows Arch 2006; 449:395-401. [PMID 16967267 ]
- Lewin K. Carcinoid tumors and the mixed (composite) glandular-endocrine cell carcinomas. Am J SurgPathol 1987;11(Suppl 1):71-86. [PMID 3544888 ]
- Capella C, La Rosa S, Uccella S, Billo P, Cornaggia M. Mixed endocrine-exocrine tumors of the gastrointestinal tract. SeminDiagnPathol 2000; 17:91-103. [PMID 10839609 ]
- Solcia E, Klöppel G, Sobin LH, Capella C, DeLellis RA, Heitz PU, et al. Histological Typing of Endocrine Tumours (WHO. World Health Organization. International Histological Classification of Tumours). 2nd edition. New York, USA: Springer Verlag, 2000. [ISBN 3540661697 ]
- Pecorella I, Memeo L, Ciardi A, Rotterdam H. An unusual case of colonic mixed adenoendocrine carcinoma: collision versus composite tumor. A case report and review of the literature. Ann DiagnPathol 2007; 11:285-90. [PMID 17630114 ]
- Jones MA, Griffith LM, West AB. Adenocarcinoid tumor of the periampullary region: a novel duodenal neoplasm presenting as biliary tract obstruction. Hum Pathol 1989; 20:198-200. [PMID 2914703 ]
- Burke A, Lee YK. Adenocarcinoid (goblet cell carcinoid) of the duodenum presenting as gastric outlet obstruction. Hum Pathol 1990; 21:238-9. [PMID 2307453 ]
- Shah IA, Schlageter MO, Boehm N. Composite carcinoidadenocarcinoma of ampulla of Vater. Hum Pathol 1990; 21:1188-90. [PMID 2227927 ]
- Misonou J, Kanda M, Kitagawa T, Ota T, Muto E, Nenohi M, Atsuta T. A case of coexisting malignant carcinoid tumor and adenocarcinoma in the papilla of Vater. GastroenterolJpn 1990; 25:630-5. [PMID 2227254 ]
- Williams IM, Williams NW, Stock D, Foster ME. Collision tumour of the ampulla of Vater: carcinoid and adenocarcinoma. HPB Surg 1997; 10:241-4. [PMID 9184878 ]
- Alex WR, Auerbach HE, Pezzi CM. Adenocarcinoid tumor of the ampulla of Vater. Am Surg 1998; 64:355-9. [PMID 9544149 ]
- Moncur JT, Lacy BE, Longnecker DS. Mixed acinar-endocrine carcinoma arising in the ampulla of Vater. Hum Pathol 2002; 33:449- 51. [PMID 12055683 ]
- Jensen RT, Rindi G, Arnold R, Lopes JM, Brandi ML, Bechstein WO, et al. Well-differentiated duodenal tumor/carcinoma (excluding gastrinomas). Neuroendocrinology 2006; 84:165-72. [PMID 17312376 ]
- Anlauf M, Garbrecht N, Bauersfeld J, Schmitt A, Henopp T, Komminoth P, et al. Hereditary neuroendocrine tumors of the gastroenteropancreatic system. Virchows Arch 2007; 451:S29-38. [PMID 17684762 ]
- Klöppel G, Rindi G, Anlauf M, Perren A, Komminoth P. Sitespecific biology and pathology of gastroenteropancreatic neuroendocrine tumors. Virchows Arch 2007; 451:S9-27. [PMID 17684761 ]
- Tanaka S, Yamasaki S, Matsushita H, Ozawa Y, Kurosaki A, Takeuchi K, et al. Duodenal somatostatinoma: a case report and review of 31 cases with special reference to the relationship between tumor size and metastasis. PatholInt 2000; 50:146-52. [PMID 10792774 ]
- Cappelli C, Agosti B, Braga M, Cumetti D, Gandossi E, Rizzoni D, AgabitiRosei E. Von Recklinghausen's neurofibromatosis associated with duodenal somatostatinoma. A case report and review of the literature. Minerva Endocrinol 2004; 29:19-24. [PMID 15258554 ]
- Fendrich V, Ramaswamy A, Slater EP, Bartsch DK. Duodenal somatostatinoma associated with Von Recklinghausen's disease. J HepatobiliaryPancreatSurg 2004; 11:417-21. [PMID 15619018 ]
- Bettini R, Falconi M, Crippa S, Capelli P, Boninsegna L, Pederzoli P. Ampullarysomatostatinomas and jejunal gastrointestinal stromal tumor in a patient with Von Recklinghausen's disease. World J Gastroenterol 2007; 13:2761-3. [PMID 17569151 ]
- Sakorafas GH, Giannopoulos GA, Parasi A, Konstantoudakis G, TzanakisN,Stergiopoulos S, et al. Large somatostatin-producing endocrine carcinoma of the ampulla of vaterinassociation with GIST in a patient with von Recklinghausen's disease. Case report and review of the literature. JOP. J Pancreas (Online) 2008; 9:633-9. [PMID 18762695 ]
- Makhlouf HR, Burke AP, Sobin LH. Carcinoid tumors of the ampulla of Vater: a comparison with duodenal carcinoid tumors. Cancer 1999; 85:1241-9. [PMID 1018912 ]
- Basile U, Cavallaro G, Polistena A, Giustini S, Orlando G, Cotesta D, et al. Gastrointestinal and Retroperitoneal Manifestations of Type 1 Neurofibromatosis. J GastrointestSurg 2009; 3. [PMID 19495890 ]
- Dayal Y, Tallberg KA, Nunnemacher G, DeLellis RA, Wolfe HJ. Duodenal carcinoids in patients with and without neurofibromatosis. A comparative study. Am J SurgPathol 1986;10:348-57. [PMID 2422964 ]
- Stewart DR, Corless CL, Rubin BP, Heinrich MC, Messiaen LM, Kessler LJ, et al. Mitotic recombination as evidence of alternative pathogenesis of gastrointestinal stromal tumours in neurofibromatosis type 1. J Med Genet 2007; 44:e61. [PMID 17209131 ]
- Maertens O, Prenen H, Debiec-Rychter M, Wozniak A, Sciot R, Pauwels P, et al. Molecular pathogenesis of multiple gastrointestinal stromal tumors in NF1 patients. Hum Mol Genet 2006; 15:1015-23. [PMID 16461335 ]
- Chetty R, Vajpeyi R. Vasculopathic changes, a somatostatinproducing neuroendocrine carcinoma and a jejunal gastrointestinal stromal tumor in apatient with type 1 neurofibromatosis. EndocrPathol 2009; 20:177-81. [PMID 19488862 ]
- Barahona-Garrido J, Aguirre-Gutiérrez R, Gutiérrez-Manjarrez JI, Tellez-Avila FI, Lopez-Arce G, Fomperoza-Torres A, et al. Association of GIST and somatostatinoma in a patient with type-1 neurofibromatosis: is there a common pathway? Am J Gastroenterol 2009; 104:797-9. [PMID 19223891 ]
- Klein A, Clemens J, Cameron J. Periampullary neoplasms in von Recklinghausen's disease. Surgery 1989; 10:815-9. [PMID 2510333 ]
- Costi R, Caruana P, Sarli L, Violi V, Roncoroni L, Bordi C. Ampullary adenocarcinoma in neurofibromatosis type 1. Case report and literature review. Mod Pathol 2001; 14:1169-74. [PMID 11706080 ]
- Nesi G, Marcucci T, Rubio CA, Brandi ML, Tonelli F. Somatostatinoma: clinico-pathological features of three cases and literature reviewed. J GastroenterolHepatol 2008;23:521-6. [PMID 17645474 ]



