Keywords
Cell Proliferation; Diabetes Mellitus, Type 2; Glucagon-Secreting Cells; Hyperlipidemias; Insulin-Secreting Cells; Islets of Langerhans
Abbreviations
ANOVA: analysis of variance; FFA: free fatty acid; GSIS: glucose-stimulated insulin secretion; HF: high fat; HFD: high fat diet; HOMA-B: homeostasis model assessmentbeta cell function; HOMA-IR: homeostasis model assessmentinsulin resistance
INTRODUCTION
Islet cells develop throughout life and demonstrate plasticity in response to metabolic demand to maintain glucose homeostasis. In mice, the pancreatic bud forms at ~e9. 5 during the primary transition comprising cells that can give rise to all three major lineages of the adult pancreas: endocrine, exocrine and duct [1]. The mesenchyme is required for the correct patterning, growth and differentiation of the embryonic pancreas [2]. In rodents, most islets develop prior to birth and during the immediate period after birth while undergoing substantial remodeling. The critical periods of islet development in rodents are islet morphogenesis at e17- 19 [3] and the first week of life when neogenesis occurs [4]. Any disturbance in the environment of the islet cells at a specific developmental time point may perturb the balance of controlling factors, thereby contributing to beta cell dysfunction and diabetes later in life [5].
An altered metabolic state of increased insulin demand, such as obesity which is strongly associated with insulin resistance, confers susceptibility to develop metabolic disease. Chronic high fat (HF) feeding is associated with the pathogenesis of obesity, insulin resistance and beta cell dysfunction. Increased free fatty acid (FFA) or saturated fatty acid influx from a high fat diet (HFD) may induce adipogenesis and metabolic syndrome, also modulating the inflammatory response [2, 3]. Rodents on chronic HFDs were obese, hyperlipidemic, glucose intolerant and insulin insensitive with impaired insulin signaling reflecting insulin resistance [6]. In another study, mice fed chronic HFDs displayed beta cell dysfunction, impaired glucose-stimulated insulin secretion (GSIS) and glucose intolerance as a consequence of insulin resistance [7].
Programming is the exposure to stimuli or insults (i. e. , events) during critical life stages, particularly during fetal and lactational life, that shape progeny metabolism and physiology immediately, transiently and/or permanently. High saturated fat programming (hereafter HF programming) is the exposure to a HFD (≥40% of mainly saturated fat as energy) during critical life stages. We previously demonstrated that HF programming altered islet cell development and physiology, specifically compromising beta cell integrity and function, in neonate [8, 9], weanling [10, 11, 12] and adolescent rats [13].
However, our recent study in the adolescent rats [13] focused on the gender-specific effects and not on global representative effects of combined genders as in the previous neonate and weanling studies. Therefore the present study will assess the islet responses to HF programming in these adolescent progeny (combined gender phenotypes). The changes in the metabolic parameters (body weight, leptinemia, glycemia, insulinemia, HOMA and lipidemia) and islet morphometry (islet cell number, size, volume, ratios and proliferation) of these adolescent progeny will be presented.
MATERIALS AND METHODS
Experimental Design
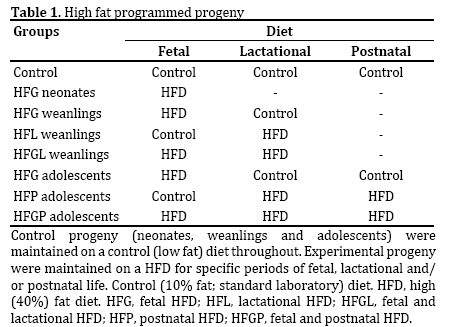
Progeny were generated from female Wistar rats after obtaining ethical approval according to previous protocols [8, 10]. The experimental groups are listed in Table 1 and refer to neonates euthanized at postnatal day 1, weanlings at postnatal day 21 and adolescents at postnatal day 90. Further, the periods of HFD maintenance are reflected in Table 1. The control progeny were maintained on a standard laboratory diet throughout fetal life (neonates) and both fetal and postnatal life (weanlings and adolescents) (Table 1). HFG adolescents were maintained on a fetal HFD, HFP adolescents on a postnatal HFD, and HFGP adolescents on a HFD throughout fetal and postnatal life (Table 1). HFG neonates, HFG, HFL and HFGL weanlings were also studied. In the weanlings, the nomenclature for HFP and HFGP adolescents were replaced by HFL and HFGL respectively where L refers to lactation as P refers to postnatal in adolescents (Table 1). The standard laboratory (control or low fat) diet comprised 10% fat, 15% protein and 75% carbohydrate (2. 6 kcal/g) as energy whereas the HFD contained 40% fat, 14% protein and 46% carbohydrate (2. 06 kcal/g). The HFD mainly comprised saturated fat and was used in previous islet studies [8, 11, 13].
Metabolic Parameters
The metabolic parameters assessed were body weight (previously determined in neonates and weanlings), pancreas weight (also adjusted for body weight: pancreas weight/body weight X 100), fasting serum leptin (rat leptin RIA kit, Linco Research, St. Charles, MO, USA; not determined in neonates due to insufficient sera volumes), blood glucose (glucometer, Precision QID, MediSense, Oxfordshire, UK) and serum insulin concentrations (rat insulin RIA kit, Linco Research; previously determined in neonates and weanlings), HOMA-insulin resistance (IR) ([fasting blood glucose (mmol/L) X fasting serum insulin (mU/L)]/22. 5)) and HOMA-beta cell function (B) ([20 X fasting insulin (mU/L)]/glucose (mmol/L) – 3. 5)). HOMA indices were adapted for neonates as sera were pooled for insulin measurements; therefore no individual insulin concentrations were available. However, the insulin was matched to glucose concentrations per group for estimation. In adolescents (not determined in neonates or weanlings), fasting serum triglyceride (GPO-PAP method, autohumalyzer A5, Human Diagnostics, Wiesbaden, Germany), total, LDL and HDL cholesterol (rapid thin-layer chromatographic-gas-liquid procedure) were determined as previously described [14]. Further, total cholesterol to HDL cholesterol ratios were determined.
For body weights, n=44 for control, n=9 for HFG, n=20 for HFP and n=10 for HFGP adolescents. For pancreas weights, n=6 for control and n=18 for HFG neonates; n=14 for control and n=6 for HFG, HFL and HFGL weanlings; n=40 for control, n=3 for HFG, n=6 for HFP and n=4 for HFGP adolescents. For leptin, n=8 for control, n=5 for HFG, n=8 for HFL and n=6 for HFGL weanlings; n=16 for control, n=6 for HFG, n=14 for HFP and n=5 for HFGP adolescents. For glucose concentrations, n=46 for control, n=13 for HFG, n=24 for HFP and n=10 for HFGP adolescents. For insulin concentrations, n=34 for control, n=8 for HFG, n=20 for HFP and n=5 for HFGP adolescents. For HOMA-IR and HOMA-B, n=10 for control and n=6 for HFG neonates; n=25 for control, n=6 for HFG, n=9 for HFL and n=5 for HFGL weanlings; n=22 for control, n=7 for HFG, n=14 for HFP (for HOMA-IR but n=13 for HOMA-B) and n=5 for HFGP adolescents. For lipid profiles, n=6 for control and HFG adolescents and n=4 for HFGP while HFP adolescents were excluded due to n=2.
Islet Morphometry
Pancreata were double immunolabeled with insulin and glucagon followed by image analysis as previously described [8]. Beta and alpha cell numbers, sizes and volumes, beta to alpha cell ratios (previously determined in neonates and HFG weanlings), alpha to beta cell ratios, islet and acinar cell proliferation indices were calculated [8, 11, 13].
Whole pancreata were harvested with 4 μm2 serial sections selected for immunolabeling experiments. Alpha cells were first immunolabeled with a polyclonal glucagon antibody (Dako, Carpinteria, CA, USA) followed by immunolabeling of the beta cells with a monoclonal insulin antibody (Sigma ImmunoChemicals, St Louis, MO, USA) [13]. For image analysis, a Canon Powershot S40 digital camera (Canon, Tochigi, Japan) mounted on an Olympus BX60 light microscope (Olympus, Tokyo, Japan) attached to a personal computer was used to capture images [13]. The final digitized images were 768 X 1024 pixels [13].
An islet was defined as a cluster of ≥8 islet cells. Image analysis was performed with the Leica Qwin Plus Software (Leica, Cambridge, UK) [13]. Total tissue area was determined by adding the tissue measured in each field of view using the interactive measurement option of the Leica software [13]. Total islet area, total beta cell area and total alpha cell area were determined by color segmentation and thresholding [13]. Beta cell number was assessed by counting the number of beta cell nuclei. Beta cell size was calculated by dividing the measured beta cell area by the number of beta cell nuclei counted and expressed in μm2 [13]. The relative beta cell volume was obtained by calculating the ratio between the area obtained by immunoreactive beta cells and the area occupied by total islet cells [13]. The same procedures were applied to estimate alpha cell number, size and volume [13]. For islet cell studies, n=8 for control, n=6 for HFG, n=7 for HFP and n=5 for HFGP adolescents. The ratio of beta to alpha cell area (i. e. , beta to alpha cell ratio) was determined by dividing the total beta cell area by the total alpha cell area [13]. The inverse calculations were applied to determine the alpha to beta cell ratio [13]. For alpha to beta cell ratios, n=6 for control and n=11 for HFG neonates; n=8 for control, n=6 for HFG, n=7 for HFL and n=6 for HFGL weanlings.
Proliferation indices of islet and acinar cells were calculated after immunostaining with the proliferation marker, Ki67 (MIB5; DakoCytomation, Glostrup, Denmark) [13]. Five hundred nuclei of each cell type (islet and acinar cells) were counted [13]. The number of proliferative cells was divided by the total number of cells to determine the islet and acinar cell proliferation indices [13]. For islet and acinar cell proliferation, n=6 for control and n=12 for HFG neonates; n=8 for control, n=6 for HFG and HFGL, and n=8 for HFL weanlings; n=6 for control and n=5 for HFG, HFP and HFGP adolescents.
STATISTICS
For neonates, the unpaired student’s t test was applied to analyze the two groups. For weanlings and adolescents, four groups were compared using one-way ANOVA followed by Bonferroni’s post-hoc test. Data are reported as means ± SEM with significance established at p<0. 05.
RESULTS
Metabolic Parameters
In neonates, leptin concentrations were not determined due to insufficient sample volumes. There were no differences in weanling leptin concentrations (Table 2). HFGP adolescents were heavier than all, whereas HFP adolescents were heavier than the control adolescents with elevated leptin concentrations in HFP and HFGP adolescents relative to control and HFG adolescents (Table 2).

HFG neonates had increased HOMA-IR with no changes in HOMA-B, despite a 55% reduction (Table 2). Weanling HOMA-IR and HOMA-B were unaltered (Table 2). Surprisingly the 88% and 74% reduction in HOMA-B in HFG and HFL weanlings respectively was not significant. HFP adolescents were hyperglycemic compared to control and HFG adolescents (Table 2). Both HFP and HFGP adolescents were hyperinsulinemic compared to control and HFG adolescents which was supported by elevated HOMA-IR indices (Table 2). There were no significant differences in HOMA-B, despite an 83% reduction in HFG adolescents compared to control adolescents (Table 2).
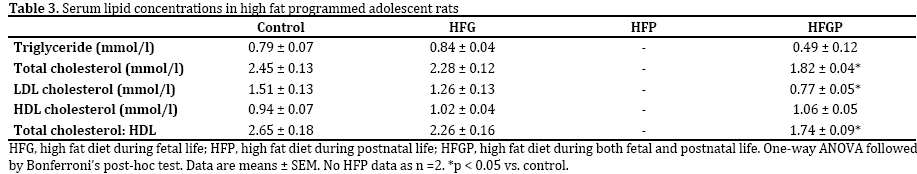
In HFGP adolescents, total and LDL cholesterol concentrations and the total cholesterol to HDL cholesterol ratios, which predicts the risk for developing atherosclerosis, were reduced compared to control adolescents (Table 3). Triglyceride and HDL cholesterol concentrations did not differ amongst the groups (Table 3).
Islet Morphology
Pancreas and Pancreas: Body Weight
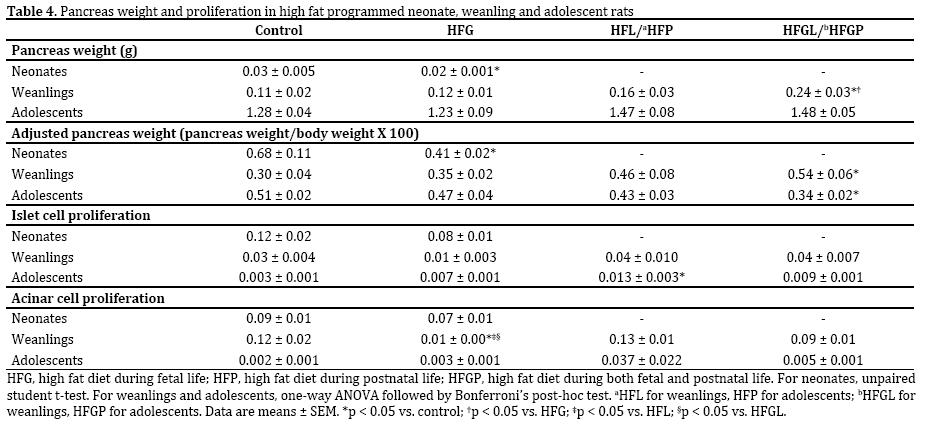
Pancreas (p=0. 0018) and pancreas:body (p=0. 0007) weights were reduced in HFG neonates compared to control neonates (Table 4). Pancreata were heavier in HFGL weanlings relative to control and HFG weanlings with pancreas:body weight increased relative to control weanlings (Table 4). In adolescents, pancreas weights did not differ; however, pancreas: body weight was reduced in HFGP adolescents compared to control adolescents (Table 4).
Islet Cell Number, Size, Volume, Ratios and Proliferation (Islet and Acinar Cells)
There were no changes in beta cell morphology in HFL and HFGL weanlings compared to control weanlings (data not shown). HFP adolescents displayed beta cell hypoplasia compared to HFGP adolescents (Figure 1A) and beta cell hypertrophy compared to control, HFG and HFGP adolescents (Figure 1B). Beta cell volume was similar amongst the groups (Figure 1C).
Figure 1(A). Beta cell number in adolescent rats maintained on a high
fat diet during fetal and/or postnatal life.
Figure 1(B). Beta cell size in adolescent rats maintained on a high fat
diet during fetal and/or postnatal life.
Figure 1(C). Beta cell volume in adolescent rats maintained on a high fat
diet during fetal and/or postnatal life.
There were no changes in alpha cell morphology in HFL and HFGL weanlings compared to control weanlings (data not shown). HFP adolescents displayed alpha cell hyperplasia relative to HFGP adolescents (Figure 2A). Alpha cell sizes (Figure 2B) and volumes (Figure 2C) were similar amongst the groups.
Figure 2(A). Alpha cell number in adolescent rats maintained on a high
fat diet during fetal and/or postnatal life.
Figure 2(B). Alpha cell size in adolescent rats maintained on a high fat
diet during fetal and/or postnatal life.
Figure 2(C). Alpha cell volume in adolescent rats maintained on a high
fat diet during fetal and/or postnatal life.
Alpha to beta cell ratios were 0. 20 ± 0. 05 in control and 0. 43 ± 0. 11 in HFG neonates with no differences. There were no changes in beta to alpha cell ratios in HFL and HFGL weanlings (data not shown). Alpha to beta cell ratios were 0. 43 ± 0. 04 in control; 0. 61 ± 0. 08 in HFG; 0. 58 ± 0. 08 in HFL and 0. 60 ± 0. 08 in HFGL weanlings with no differences. The beta to alpha cell (Figure 3A) and alpha to beta cell (Figure 3B) ratios were not altered in adolescents.
Figure 3(A). Islet cell ratios. Beta to alpha cell ratio in adolescent rats
maintained on a high fat diet during fetal and/or postnatal life.
Figure 3(B). Islet cell ratios. Alpha to beta cell ratio in adolescent rats
maintained on a high fat diet during fetal and/or postnatal life.
There were no differences in either islet or acinar cell proliferation indices in neonates (Table 4). Acinar cell proliferation indices were reduced in HFG weanlings compared to control, HFL and HFGL weanlings with no differences in the islet cell proliferation indices (Table 4). Islet cell proliferation indices were increased in HFP compared to control adolescents; there were no differences in the acinar cell proliferation indices (Table 4).
DISCUSSION
The present study focused on HF programming of metabolic and islet parameters in adolescent Wistar rats. Specifically we investigated the changes in the metabolic parameters (body weight, leptinemia, glycemia, insulinemia, HOMA and lipidemia) and islet morphometry (islet cell number, size, volume, ratio and proliferation) in these progeny. Further, we sought to combine new data to previous data in neonate and weanling progeny. This allowed standardization of metabolic and islet parameters in neonate, weanling and adolescent progeny.
Neonates
The HFG neonates were hyperglycemic [8] with impaired insulin release from islets [9] and insulin resistant (present study). Further, beta cell numbers and volumes were diminished whereas alpha cell numbers, sizes and volumes were augmented [8]; this diabetogenic phenotype demonstrated the immediate detrimental fetal.
HF programming effects on islet architecture and beta cell function. The beta cell dysfunction and insulin resistance appears in response to hyperglycemia. Hyperglycemia in utero programs the endocrine pancreas [15]; in HFG neonates this induced beta cell dysfunction and insulin resistance.
The capacity of beta cells to proliferate in response to insulin resistance is critical for glucose homeostasis and preventing the onset of type 2 diabetes [16]. Beta cell proliferation requires co-staining with a beta cell marker (i. e. , insulin) and a proliferation marker (e. g. , Ki67). In the present study, islet and acinar cell proliferation did not discern the cell subtypes. With no increases in islet or acinar cell proliferation in HFG neonates, no new beta cells were available to sustain beta cell compensation in response to increased insulin demand. This likely contributed to the compromised beta cell development (non-replenishment of beta cells) and function. An increase in alpha cell number and glucagon secretion may exacerbate insulin resistance [17]. The insulin resistant HFG neonates had alpha cell hyperplasia and hypertrophy resulting in increased alpha cell volume [8] that likely exacerbated their insulin resistance.
Weanlings
Various weanling phenotypes presented after HF programming during different developmental stages. HFG weanlings, with fetal HF maintenance, had low body weights, hypoinsulinemia [10] and glucose intolerance [12] with normal islet morphology [10] but reduced acinar cell proliferation (present study). Acinar and ductal cells co-inhabit the pancreas to form the majority of this microorgan. With a similar lineage during ontogeny and their close proximity during maturity, the trans-differentiation of these non-islet cells to beta cells is attractive. The combination of the transcription factors MafA, Pdx1 and Ngn3 was reported to drive the reprogramming of acinar cells to islet cells in the pancreas of adolescent immune-compromised mice [18]. The reduced acinar cell proliferation in HFG weanlings presented independent of changes in islet cell proliferation and morphology. The low fat diet intervention, during the critical period of pancreatic remodeling from birth to weaning, improved islet outcomes [10] but as a consequence resulted in reduced acinar cell proliferation. This may constrain trans-differentiation should insulin demand increase. The contribution and dynamics of various pancreatic cell types to maintain glucose homeostasis requires further investigation.
HFL weanlings, with HF maintenance from birth to 3 weeks of age, were hyperglycemic [10] and the most glucose intolerant [12], despite normal islet morphology (present study). This revealed that lactational HF programming (i. e. , HF maintenance during lactational life) induced a diabetogenic phenotype characterized by hyperglycemia and glucose intolerance but independent of altered islet morphology and insulin resistance.
HFGL weanlings, maintained on a HFD throughout fetal life and the first 3 weeks of life, had increased body weights, hypoinsulinemia [10] and glucose intolerance [12] independent of altered islet morphology and insulin resistance (present study). In HFGL weanlings, the chronic HFD maintenance may result in the accumulation of ectopic pancreatic fat thereby increasing pancreatic weight. Ectopic pancreatic fat impairs GSIS [19] which may contribute to the hypoinsulinemia and glucose intolerance in HFGL weanlings clearly reflecting beta cell dysfunction.
The metabolic changes in HFG, HFL and HFGL weanlings were independent of changes in islet morphometry. New beta cells can be formed either by proliferation of existing beta cells (replication) or by de novo differentiation from non-beta, viz. , pancreatic (i. e. , non-beta islet cells, acinar and duct cells) and extra-pancreatic cells (neogenesis). Beta cells undergo active proliferation and apoptosis during the first 20 days of life which coincides with the lifespan of these weanlings, also associated with a transiently reduced islet growth rate in rodents [20]. Lactation is a critical period for beta cell maturation [21]. Islet remodeling during lactational life may help to maintain normal islet morphology, despite the HF maintenance in HFL and HFGL weanlings. In HFG weanlings, the low fat (control) diet during lactational life prompts the restoration of compromised beta cell morphology that presented at birth. Lactation may hold the greatest beta cell compensatory capacity in progeny given that extensive islet remodeling occurs and may equip beta cells to correct prior insults. Healthy nutrition during lactational life therefore improves progeny health outcomes.
Adolescents
Like the weanlings, the adolescents also presented different phenotypes shaped by the specific period of HF maintenance. Surprisingly, HFG adolescents displayed normal metabolic and islet parameters mimicking the control adolescent phenotype. Therefore the adverse fetal HF programming effects on islet morphology and function were corrected by chronic low fat maintenance throughout postnatal life. Beta cells are dynamic and the life stage influences beta cell turnover. Compensatory beta cell expansion (i. e. , hyperplasia and hypertrophy in response to increased insulin demand) is limited and declines with age. In Wistar rats, beta cell function decreases with age limiting their compensatory capacity which may contribute to the development of diabetes [22]. Therefore, in HFG adolescents, the adverse diabetogenic phenotype that presented at the neonatal life stage may recur upon ageing as their beta cell compensatory capacity diminishes.
The HFP adolescents demonstrated the most adverse metabolic and islet changes characterized by increased body weights, hyperleptinemia, hyperglycemia, hyperinsulinemia and insulin resistance with beta cell hypertrophy and increased islet cell proliferation. HF maintenance from birth to adolescence therefore induced a mildly obese but severe diabetogenic phenotype. The enlarged beta cells in HFP adolescents appeared incapable of maintaining glucose homeostasis and therefore did not ameliorate their diabetogenic state. Beta cell hyperplasia is required, and despite increased islet cell proliferation, neither beta nor alpha cell number increased suggesting that other islet cell types, viz. , delta, PP and/or epsilon cells may be proliferating. Therefore non-beta and alpha islet cells may present an intra-islet source for replenishing beta cells. The ability of an organism to maintain its beta cell mass during adulthood is critical for achieving glucose homeostasis and preventing diabetes [23]. In HFP adolescents, the partial beta cell compensation comprises beta cell hypertrophy and hyperinsulinemia to counter insulin resistance which was insufficient to restore glucose homeostasis.
The beta and alpha cell relationship is critical considering that insulin and glucagon normally partner to maintain circulating glucose concentrations within a tight physiological range [24]. Interestingly, HFGP adolescents demonstrated beta cell hyperplasia and alpha cell hypoplasia relative to HFP adolescents reflecting altered islet cell numbers. Programming of the endocrine pancreas ultimately originates from hyperglycemia during fetal and/or lactational life [15]. Since HFGP adolescents were exposed to maternal hyperglycemia as fetuses and were hyperglycemic as neonates unlike HFP adolescents [8], hyperglycemia during fetal life appeared to shift islet cell numbers.
HFGP adolescents were heaviest, most hyperleptinemic, hyperinsulinemic and insulin resistant reflecting a severe obese, insulin resistant state. The combined effects of HF programming (during fetal and lactational life) with chronic HFD maintenance (from weaning to adolescence) therefore induced obesity. Further, the hyperleptinemia, hyperinsulinemia and insulin resistance in HFGP adolescents was also relative to the HFG adolescents, reinforcing that HFG adolescents mimicked the control phenotype at 3 months of age. In diabetes and obesity, obesity-associated insulin resistance causes hyperinsulinemia as the beta cells are hyperstimulated to release more insulin, although the physiological mechanism for this hyperstimulation remains unknown since it often occurs prior to hyperglycemia [25]. In obeseinsulin resistant HFGP adolescents, these metabolic traits manifest in the absence of hyperglycemia signaling a prediabetic state.
Dyslipidemia is characterized by increased circulating triglyceride, increased LDL and reduced HDL concentrations. Mature Wistar rats showed dyslipidemia reflected by increased serum triglyceride, total cholesterol and LDL cholesterol concentrations [26]. However, HDL cholesterol concentrations were also increased [26] due to rats mainly carrying cholesterol as HDL. Despite a severe obese hyperleptinemic and insulin resistant state, HFGP adolescents appeared to have a favorable cardiometabolic profile given their reduced total and LDL cholesterol and total cholesterol to HDL ratios which was conflicting. The HFGP adolescents may be in an adaptive metabolic state to adjust to the chronic HFD maintenance. In mice fed a HFD, there was a 20-fold increase in pancreatic triglycerides [27]. No increase in circulating triglycerides was found in the HF programmed adolescents. Considering the obese state of HFP and HFGP adolescents, determining the pancreatic triglyceride and fat content should be investigated given their direct association with beta cell dysfunction and insulin resistance.
Perspectives on Insulin Resistance, Beta Cell Compensation and Dysfunction
The association of insulin resistance and beta cell dysfunction in the pathogenesis of diabetes is complex and depends on the stage and severity of the disease. Some insulin resistant individuals do not develop diabetes; diabetes only manifests if beta cells fail to synthesize and secrete sufficient insulin [28]. Beta cells initially compensate for the insulin resistance associated with obesity by increasing insulin secretion [29]. HFG neonates displayed insulin resistance and beta cell dysfunction (hyperglycemia [8], reduced GSIS [9] and reduced HOMA-B albeit non-significant). Therefore insulin resistance and beta cell dysfunction co-presented in the same diabetogenic phenotype. In HFG neonates and HFP adolescents, insulin insensitivity may be advanced as glycemic disturbances were already evident. Prolonged hyperinsulinemia desensitizes insulin receptors and causes insulin resistance which burdens and diminishes beta cell function thereby promoting the onset of diabetes [30]. This links beta cell dysfunction and insulin resistance. With beta cell dysfunction and insulin co-presenting and their robust association in the pathogenesis of type 2 diabetes, both diabetogenic states should be simultaneously targeted to prevent the onset of overt diabetes.
In HFP and HFGP adolescents, the increase in body weight and hyperleptinemia reflected obesity supported by their hyperinsulinemic insulin resistant state. In HFP adolescents, hyperinsulinemia may be in response to hyperglycemia and/or insulin resistance. This partial beta cell compensatory response is inadequate for restoring glucose homeostasis therefore insulin resistance manifests. In HFGP adolescents, hyperinsulinemia appears to be in response to insulin resistance as hyperglycemia had not yet manifested. Further, in HFGP adolescents, with no changes in beta cell morphology or glycemia, beta cell function appears somewhat intact, although compensatory hyperfunction is reflected by hyperinsulinemia.
HOMA-B indices were aberrant at all life stages. Despite large reductions of 55-88% in HOMA-B indices in HF programmed progeny (neonates, weanlings and adolescents) without significance established; beta cell dysfunction was inferred. This lack of significance reflected variances in glucose and insulin concentrations suggesting that the equation may require refinement to minimize the inherent variability of glycemia and insulinemia that is inevitably compounded when factored into the equation. Reducing the glycemic and insulinemic variability by increasing sample sizes and factoring in an adjustment to counter the variability may reinforce HOMA-B. Also, a specialized formula adapted for rodents should enhance accuracy. Finally, HOMA-B should be supplementary to GSIS data to better conclude and confirm the beta cell functional state. In contrast, the HOMA-IR formula appeared more robust, given that differences were established with the same input data.
CONCLUSION
HF programming induces metabolic and islet derangements inducing various diabetogenic phenotypes at different life stages. Fetal HF programming resulted in an immediate diabetogenic phenotype at birth that improved at weaning and mimicked the control phenotype at adolescence. In adolescents, postnatal HF maintenance induced the most severe diabetogenic phenotype. Continuous HF maintenance, throughout fetal life up to 3 months of age, resulted in obese, hyperleptinemic and insulin resistant adolescents. The postnatal period from birth to adolescence represents an extension of HF programming of islet cells and metabolic disease.
Conflict of Interest
There was no conflict of interest among authors.
Acknowledgements
The authors are also grateful to employees of the Diabetes Discovery Platform, Primate Unit and Nutrition Intervention Research Unit of the South African Medical Research Council for technical assistance.
References
- Carolan PJ, Melton DA. New findings in pancreatic and intestinal endocrine development to advance regenerative medicine. Curr Opin Endocrinol Diabetes Obes 2013; 20:1-7. [PMID:23249759]
- Gittes GK. Developmental biology of the pancreas: a comprehensive review. Dev Biol 2009; 326:4-35. [PMID:19013144]
- Miralles F, Battelino T, Czernichow P, Scharfmann R. TGF-beta plays a key role in morphogenesis of the pancreatic islets of Langerhans by controlling the activity of the matrix metallo proteinase MMP-2. J Cell Biol 1998; 143:827-836. [PMID:9813100]
- Kaung HL. Growth dynamics of pancreatic islet cell populations during fetal and neonatal development of the rat. Dev Dyn 1994; 200:163-175. [PMID:7919502]
- Reusens B, Theys N, Dumortier O, Goosse K, Remacle C. Maternal malnutrition programs the endocrine pancreas in progeny. Am J Clin Nutr 2011; 94:1824S-1829S. [PMID:21562089]
- Wu Y, Wu T, Wu J, Zhao L, Li Q, et al. Chronic inflammation exacerbates glucose metabolism disorders in C57BL/6J mice fed with high-fat diet. J Endocrinol 2013; 219:195-204. [PMID:24029730]
- Collins SC, Hoppa MB, Walker JN, Amisten S, Abdulkhader F, Bengtsson M, et al. Progression of diet-induced diabetes in C57Bl6J mice involves functional dissociation of Ca2+ channels from secretory vesicles. Diabetes 2010; 59:1192-1201. [PMID:20150285]
- Cerf ME, Williams K, Nkomo XI, Muller CJ, Du Toit DF, Louw J, et al. Islet cell response in the neonatal rat after exposure to a high-fat diet during pregnancy. Am J Physiol RegulIntegr Comp Physiol 2005; 288:R1122-R1128. [PMID:15705804]
- Cerf ME, Chapman CS, Muller CJ, Louw J. Gestational high-fat programming impairs insulin release and reduces Pdx-1 and glucokinaseimmunoreactivity in neonatal Wistar rats. Metabolism 2009; 58:1787- 1792. [PMID:19604517]
- Cerf ME, Muller CJ, Du Toit DF, Louw J, Wolfe-Coote SA. Hyperglycaemia and reduced glucokinase expression in weanling offspring from dams maintained on a high-fat diet. Br J Nutr 2006; 95:391-396. [PMID:16469158]
- Cerf ME, Williams K, Chapman CS, Louw J. Compromised beta-cell development and beta-cell dysfunction in weanling offspring from dams maintained on a high-fat diet during gestation. Pancreas 2007; 34:347- 353. [PMID:17414058]
- Cerf ME, Louw J. High fat programming induces glucose intolerance in weanling Wistar rats. Horm Metab Res 2010; 42:307-310. [PMID:20195946]
- Cerf ME, Chapman CS, Louw J. High-fat programming of hyperglycemia, hyperinsulinemia, insulin resistance, hyperleptinemia, and altered islet architecture in 3-month-old wistar rats. ISRN Endocrinol 2012; 2012:627270. [PMID:22988521]
- Tichelaar HY, SpinnlerBenade AJ, Daubitzer AK, Kotze TJ. An improved rapid thin-layer chromatographic-gas-liquid chromatographic procedure for the determination of free fatty acids in plasma. Clin Chim Acta 1989; 183:207-215. [PMID:2791305]
- Portha B, Chavey A, Movassat J. Early-life origins of type 2 diabetes: fetal programming of the beta-cell mass. Exp Diabetes Res 2011; 2011:105076. [PMID:22110471]
- Blandino-Rosano M, Alejandro EU, Sathyamurthy A, Scheys JO, Gregg B, Chen AY, et al. Enhanced beta cell proliferation in mice overexpressing a constitutively active form of Akt and one allele of p21Cip. Diabetologia 2012; 55:1380-1389. [PMID:22327314]
- Dunning BE, Gerich JE. The role of alpha-cell dysregulation in fasting and postprandial hyperglycemia in type 2 diabetes and therapeutic implications. Endocr Rev 2007; 28:253-283. [PMID:17409288]
- Zhou Q, Brown J, Kanarek A, Rajagopal J, Melton DA. In vivo reprogramming of adult pancreatic exocrine cells to beta-cells. Nature 2008; 455:627-632. [PMID:18754011]
- Tushuizen ME, Bunck MC, Pouwels PJ, Bontemps S, van Waesberghe JH, Schindhelm RK, et al. Pancreatic fat content and beta-cell function in men with and without type 2 diabetes. Diabetes Care 2007; 30:2916- 2921. [PMID:17666465]
- Scaglia L, Cahill CJ, Finegood DT, Bonner-Weir S. Apoptosis participates in the remodeling of the endocrine pancreas in the neonatal rat. Endocrinology 1997; 138:1736-1741. [PMID:9075738]
- Bouwens L, Rooman I. Regulation of pancreatic beta-cell mass. Physiol Rev 2005; 85:1255-1270. [PMID:16183912]
- Liu Y, Shi S, Gu Z, Du Y, Liu M, Yan S, et al. Impaired autophagic function in rat islets with aging. Age (Dordr) 2013; 35:1531-1544. [PMID:22843415]
- Ackermann AM, Gannon M. Molecular regulation of pancreatic beta-cell mass development, maintenance, and expansion. J Mol Endocrinol 2007; 38:193-206. [PMID:17293440]
- Unger RH, Orci L. Paracrinology of islets and the paracrinopathy of diabetes. Proc Natl Acad Sci USA 2010; 107:16009-16012. [PMID:20798346]
- Mehran AE, Templeman NM, Brigidi GS, Lim GE, Chu KY, Hu X, et al. Hyperinsulinemia drives diet-induced obesity independently of brain insulin production. Cell Metab 2012; 16:723-737. [PMID:23217255]
- Ghezzi AC, Cambri LT, Botezelli JD, Ribeiro C, Dalia RA, Rostom de Mello MA. Metabolic syndrome markers in wistar rats of different ages. Diabetol Metab Syndr 2012; 4:16. [PMID:23249759]
- Hoppa MB, Collins S, Ramracheya R, Hodson L, Amisten S, Zhang Q, et al. Chronic palmitate exposure inhibits insulin secretion by dissociation of Ca(2+) channels from secretory granules. Cell Metab 2009; 10:455- 465. [PMID:19945403]
- Weir GC, Bonner-Weir S. Islet beta cell mass in diabetes and how it relates to function, birth, and death. Ann NY Acad Sci 2013; 1281:92-105. [PMID:23363033]
- Kasuga M. Insulin resistance and pancreatic beta cell failure. J Clin Invest 2006; 116:1756-1760. [PMID:16823472]
- Zhang Y, Xiao M, Niu G, Tan H. Mechanisms of oleic acid deterioration in insulin secretion: role in the pathogenesis of type 2 diabetes. Life Sci 2005; 77:2071-2081. [PMID:15935394]









