Shounak Majumder1, Constantin A Dasanu2
1Department of Internal Medicine, University of Connecticut. Farmington, CT, USA.
2Department of Hematology-Oncology, Saint Francis Medical Center. Hartford, CT, USA
- *Corresponding Author:
- Shounak Majumder
Department of Internal Medicine
University of Connecticut
263 Farmington Avenue; Farmington
CT 06030; USA
Phone: +1-860.560.3862
Fax: +1-860.679.2562
E-mail: smajumder@resident.uchc.edu
Received February 13th, 2013 – Accepted April 21st, 2013
Keywords
alpha-Fetoproteins; Carcinoma, Hepatocellular; Pancreatic Neoplasms
INTRODUCTION
The term “hepatoid tumor” refers to a primary extrahepatic neoplasm that demonstrates either partial or complete histopathological resemblance to hepatocellular carcinoma [1]. Initially described in a patient with a primary gastric malignancy, this rare pathological entity was subsequently documented in lung, gastrointestinal and genitorurinary tract cancers [2]. Nonetheless, stomach remains the most common site involved with this neoplasm. Pancreatic hepatoid tumors are rare, with less than a dozen cases reported to date. We report herein a case of a metastatic hepatoid tumor of pancreas and discuss the inherent challenges associated with the diagnosis and management of this rare tumor.
CASE REPORT
A 60-year-old man presented in December 2011 with left upper quadrant pain, jaundice and nausea. Physical examination was remarkable for jaundice and moderate hepatomegaly. Computed tomography (CT) scan of abdomen demonstrated a 5.8x6.0 cm mass in the head of the pancreas and multiple hypodense lesions in the right lobe of liver, the largest measuring 2.8 cm (Figure 1). A PET-CT scan showed hypermetabolic activity in the pancreatic mass, liver lesions and peri-pancreatic lymph nodes. Serum CA 19-9 was elevated at 373 U/mL (reference range: 0-37 U/mL), but alphafetoprotein (AFP) level was within normal range. Subsequently, he underwent an ultrasound-guided biopsy of the liver. Ten days later, he presented to his physicians’ office with complaints of chills and lightheadedness, and was noted to have a blood pressure of 72/48 mmHg. At that juncture, he was diagnosed with septic shock and acute renal failure secondary to cholangitis. Endoscopic drainage was attempted but could not be performed due to duodenal infiltration by the mass. He then underwent percutaneous transhepatic biliary drainage, with a significant clinical improvement and resolution of the acute renal failure. The liver biopsy revealed a poorly differentiated malignant neoplasm with extensive focal necrosis (Figure 2). No definite histopathologic evidence of cirrhosis was identified in the non-neoplastic liver tissue of the sections examined. Immunohistochemical stains showed the tumor cells to be positive for CAM 5.2, vimentin and hepatocyte paraffin 1, and negative for AE1, AE3, CK7, CK20, alpha-fetoprotein, CD31, CEA, CA19.9, CDX2, HMB45, Melan A, and S100 (Figure 2). Histopathologic findings were initially felt to support a diagnosis of a poorly differentiated hepatocellular carcinoma. However, the clinical presentation with a locally invasive dominant pancreatic mass and innumerable small liver lesions in a non-cirrhotic liver, led to the diagnosis of metastatic hepatoid pancreatic cancer. The patient showed modest clinical improvement with biliary drainage and he received palliative chemotherapy with gemcitabine prior to the discharge from the hospital. An endoscopic ultrasound with biopsy of the pancreatic mass was planned. However, his general condition continued to decline over the next few months and he died in March 2012. An autopsy was not permitted by the family.
Figure 1. CT scan of abdomen showing a mass in the head of the
pancreas (white arrows).
Figure 2. Liver biopsy demonstrating: a. poorly differentiated
neoplasm; b. higher magnification of the poorly differentiated
carcinoma demonstrating probable evidence of bile production
by tumor cells (black arrow); c. positive staining with CAM 5.2;
and d. focal weakly positive staining with hepar-1.
DISCUSSION
The pathogenesis of hepatoid tumors is not completely understood. It has been commonly accepted that pancreas and liver share their embryologic origin from the foregut endoderm. This theory of common origin appears to be the most widely accepted explanation of the pathophysiologic basis of pancreatic hepatoid tumors. It is believed that activation of liver-specific genes during the process of carcinogenesis results in hepatocellular differentiation of the malignant pancreatic cancer cells.
A systematic English literature search revealed 13 prior reported cases of pancreatic hepatoid tumor (Table 1) [3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14]. Clinical manifestations of these rare tumors vary ranging from incidentally detected abdominal masses to painless jaundice and weight loss. The presenting manifestation may be influenced by the primary differentiation of the pancreatic tumor. The median age at diagnosis is 52.5 years (range: 28-80 years) , which is lower than the usual age distribution of pancreatic adenocarcinoma. There is a definite male preponderance (M:F ratio = 2:1), and the pancreatic head and body appear to be the most commonly affected sites. More than 70% of the previously reported cases described tumors sized greater than 5 cm at the time of initial diagnosis (range: 0.5-11 cm). Eight of the 14 cases (57%) had evidence of extra-pancreatic disease at presentation, with the liver being the commonest site of metastasis.
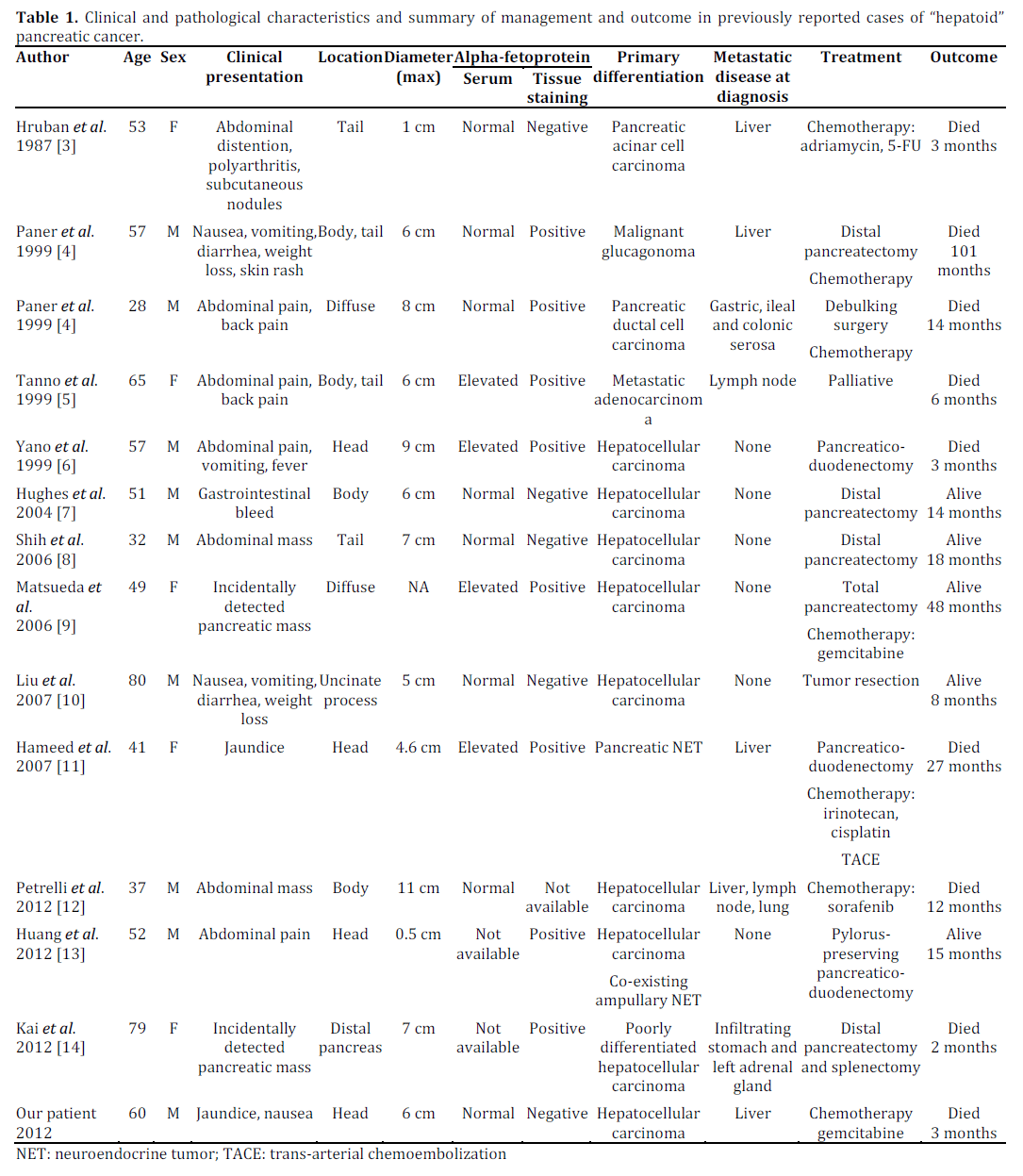
The diagnosis of hepatoid cancer is often difficult, given the lack of definite diagnostic criteria. The hepatoid neoplastic cells may appear as well-differentiated “hepatocyte-like” eosinophilic polyhedral cells with “sinusoid-like” endothelial lining and evidence of bile production or as haphazardly arranged poorly differentiated pleomorphic cells. In the latter scenario, histology is often non-diagnostic and immunohistochemistry plus clinical data are vital for making the diagnosis. Staining for AFP is a useful diagnostic test, and previous reports demonstrated positive staining with a wide variety of other immunohistochemical stains including carcinoembryonic antigen (CEA), albumin, epithelial membrane antigen, and cytokeratin (CAM 5.2). Tissue AFP staining was positive in 8/11 previously reported cases, and this may be seen in the absence of elevated serum AFP levels (Table 1). Neither serum AFP nor tissue AFP staining seems to have prognostic implications. The patient had a poorly differentiated tumor negative for CEA and AFP, but positive for CAM 5.2 and with histologic evidence of bile production by the tumor cells. Although hepar-1 positivity is more characteristic of a hepatocellular carcinoma, the histological findings combined with the clinical presentation of a dominant pancreatic mass with multiple small intrahepatic lesions led to the diagnosis of metastatic hepatoid tumor of pancreas. A pancreatic biopsy would have been ideal but could not be performed due to the patient’s rapid clinical deterioration.
Based on available evidence, the six-month and one-year mortality for hepatoid cancer are 36% (5/14) and 43% (6/14), respectively, which highlights the aggressive nature of this tumor (Table 1). Regretfully, long-term follow up data are not available in most identified literature cases. Lack of larger registry based data makes it difficult to formulate an effective management plan in these patients. Various chemotherapeutic regimens have been tried, but with little success. A recently published report describes a meaningful response to sorafenib, an oral multi-kinase inhibitor approved for hepatocellular carcinoma [12], but more clinical experience is required before any conclusions can be reached. In view of the fact that both hepatic and pancreatic carcinomas are not very responsive to chemotherapy, early diagnosis and radical surgery may provide the best opportunity for long-term cancer-free survival.
To conclude, very little is known about the hepatoid variant of pancreatic cancer and, in the absence of a standardized management approach, prognosis is grave. There is a need for increasing awareness about this rare variant of pancreatic cancer among pathologists, gastroenterologists and oncologists. It is often difficult to differentiate this unusual variant from metastatic hepatocellular cancer and clinical presentation, imaging and immunohistochemistry are essential diagnostic aids. As AFP may not be a reliable tumor marker in these cases, a well-informed pathologist and appropriate staining are essential for early diagnosis. Given the dismal outcome with traditional chemotherapy, the palliative role of other modalities such as newer agents, transarterial chemoembolization (TACE), cyber-knife and radiofrequency ablation is certainly worth exploring.
Conflict of interest
The authors do not have any conflict of interest or financial disclosures
References
- Ishikura H, Fukasawa Y, Ogasawara K, et al.An AFP-producing gastric carcinoma with feature of hepatic differentiation: a case report. Cancer 1985;56:840-8
- Metzgeroth G, Ströbel P, Baumbusch T, Reiter A, Hastka J. Hepatoid adenocarcinoma - review of the literature illustrated by a rare case originating in the peritoneal cavity. Onkologie. 2010;33:263-9.
- Hruban RH, Molina JM, Reddy MN, et al. A neoplasm with pancreatic and hepatocellular differentiation presenting with subcutaneous fat necrosis.Am J Clin Pathol. 1987;88:639-45
- Paner GP, Thompson KS, Reyes CV. Hepatoid carcinoma of the pancreas. Cancer. 2000;88:1582-9
- Tanno S, Obara T, Fujii T, et al. Alpha-Fetoprotein-producing adenocarcinoma of the pancreas presenting focal hepatoid differentiation. Int J Pancreatol. 1999;26:43-7.
- Yano T, Ishikura H, Wada T, et al. Hepatoid adenocarcinoma of the pancreas. Histopathology. 1999;35:90-2
- Hughes K, Kelty S, Martin R. Hepatoid carcinoma of the pancreas. Am Surg. 2004;70:1030-3
- Shih NN, Tsung JS, Yang AH, et al. A unique pancreatic tumor with exclusive hepatocytic differentiation. Ann Clin Lab Sci. 2006 Spring;36:216-21
- Matsueda K, Yamamoto H, Yoshida Y, et al. Hepatoid carcinoma of the pancreas producing protein induced by vitamin K absence or antagonist II (PIVKA-II) and alpha-fetoprotein (AFP). J Gastroenterol. 2006;41:1011-9
- Liu CZ, Hu SY, Wang L, et al. Hepatoid carcinoma of the pancreas: a case report. Chin Med J (Engl). 2007;120:1850-2
- Hameed O, Xu H, Saddeghi S, et al. Hepatoid carcinoma of the pancreas: a case report and literature review of a heterogeneous group of tumors. Am J Surg Pathol. 2007;31:146-52
- Petrelli F, Ghilardi M, Colombo S, et al. A rare case of metastatic pancreatic hepatoid carcinoma treated with sorafenib. J Gastrointest Cancer. 2012;43:97-102
- Huang SC, Chang HC, Yeh TS, Ng KF, Chen TC. Hepatoid microcarcinoma of the pancreas: a case report and review of the literature. Chang Gung Med J. 2012;35:285-91.
- Kai K, Nakamura J, Ide T, et al. Hepatoid carcinoma of the pancreas penetrating into the gastric cavity: a case report and literature review. Pathol Int. 2012;62:485-90.



