Keywords
Global DNA methylation; Imprinting center; SNRPN; LINE-1 methylation assay; Obesity
Introduction
Obesity characterized by a body mass index (BMI) over 30 is a pervasive global health problem that is increasing in frequency and severity [1]. Genetic predispositions for obesity combined with increasing environmental changes likely drive the increase in obesity rates with growing numbers of obese individuals living in the United States and around the world. The role of genetics in obesity can be emphasized by examining rare genetic syndromes with obesity as a core feature such as fragile X, Prader-Willi, Alström, and Bardet- Biedl syndromes [2-5]. Obesity is one of the cardinal features in Prader-Willi syndrome (PWS), a neurodevelopmental disorder due to errors in genomic imprinting usually as a result of a paternally derived deletion of the chromosome 15q11- q13 region [6-9]. Almost all individuals with PWS are considered obese, with the average age of onset of obesity occurring after 2 years of age leading to significant health problems linked to most health issues found in PWS including heart failure, type 2 diabetes, and a fatty liver [7,9].
Genomic imprinting is an important gene regulatory process emerging as a contributor to obesity and several diseaseassociated disorders. Nearly all imprinted genes have CpG-rich differentially methylated regions (DMR) typically arranged in clusters or domains on different chromosomes and under control of an imprinting center. Methylation status at these centers and nearby elements regulates gene expression often important for cellular growth, development and viability [10]. Methylation is typically associated with a repression of gene expression. Parental origin impacts methylation status with the paternal or maternal genomes exerting counteracting influences on gene expression and often involved with embryonic development and growth [11].
Approximately 1% of all mammalian genes are thought to be imprinted with about 150 imprinted genes identified and distributed across at least 115 chromosome bands within the genome [12]. The expression of imprinted genes is often tissue and stage specific. But, the monoallelic expression of an imprinted gene is not absolute and may incorporate differential transcription rates balanced between the two parental alleles [13]. Genes also appear to be clustered under the regulation of a single imprinting-controlling element which may involve higher order regulatory factors. Maternally contributed genes are usually replicated or expressed at different rates than paternal genes. Paternally expressed genes generally enhance growth of the fetus while maternally expressed genes inhibit growth. However, methylated or inactive genes are not absolute and these genes can be reactivated in male and female gametogenesis [14]. Cytosine methylation occurs in 60-80% of all mammalian somatic cell CpG dinucleotides, but most CpG islands are methylation free in the promoter regions of most active genes. Areas that are more heavily methylated (e.g., heterochromatin or repetitive sequences regions) contribute to gene silencing [15-17].
Global methylation processes are impacted by repetitive sequences that are selectively methylated and widely distributed across the genome which may have significance in the regulation of gene expression. About one-half of the human DNA sequences are noted to have repetitive elements [18]. For example, Alu repeats are short interspersed elements (SINE) that are less than 300 base pairs in size and rich in CG nucleotides at CpG islands. They are ancestral in nature, derived from small cytoplasmic RNAs and are the most abundant transposable elements in humans leading to mutations [19]. Approximately 30% of Alu sites with potential CpG islands are actual CpG sites that can be methylated. They are implicated in inherited diseases and malignancies. Long interspersed elements (LINE-1) distributed throughout the genome are rich in AT nucleotides and at locations or sites for gene regulation [20-22]. These elements constitute a significant proportion of the human genome (e.g., LINE-1 consists of 18% and Alu repeats at 10% of the genome). Changes in methylation of these elements typically correlate with global methylation patterns impacting gene expression and play a role in human development, disease and possibly obesity [23-26]. Global methylation has been linked to obesity status and weight loss in prior investigations [27,28]. An inverse relationship between global methylation and central obesity has been reported in boys that predict both obesityrelated problems (e.g., diabetes and cardiovascular disease) and obesity development [29]. A separate case report of a Korean woman found a U-shaped relationship between Alu methylation status and BMI, but a paucity of DNA methylation studies in obesity exist and even fewer studies in syndromic obesity such as PWS, a focus of our study [30].
The present study tests the hypothesis that global DNA methylation is related to obesity status and correlated with measures of known imprinted single genes (e.g., H19, GNAS) linked to obesity-related conditions and genes on chromosome 15 in the region involved in the causation of Prader-Willi syndrome, the most common known cause of life-threatening obesity in humans [31]. PWS is characterized by nearly 100% methylation of SNRPN (e.g., Rethmeyer et al., 2013 [32]), but the impact of this methylation change on regional and global methylation status is not fully characterized or understood in relationship to other imprinted genes on chromosome 15 (in cis) or other imprinted obesity-related genes on other chromosomes (in trans). We chose to examine the global methylation patterns of individuals with non-syndromic and syndromic obesity relative to lean individuals by targeting LINE-1 elements within the genome involved in gene activity and regulation. We utilized two different global methylation analysis methods and analyzed gene specific methylation patterns for imprinted, non-imprinted and obesity-related genes using targeted gene methylation methods. To further understanding and describe the methylation effects in human obesity in either cis or trans states, we compared genomic DNA isolated from whole blood specimens from a cohort of lean and obese individuals and those diagnosed with nonsyndromic obesity and PWS.
Materials and Methods
Participant Characteristics
Study participants were recruited as part of an ongoing research program on obesity-related genomics and Prader- Willi syndrome. All study procedures were performed in accordance with guidelines for the ethical conduct of research on humans with oversight from the local Institutional Review Board and written consent was obtained from all participants or their guardians. Peripheral blood samples were collected from a total of 91 adult participants [46 males, 45 females; mean age=35.1±9.2 (19-55) years] - see Table 1.
| Characteristic |
Lean, N=26 |
Obese, N=26 |
PWS, N=39 |
| Male, N (per cent) |
12 (46%) |
14 (54%) |
20 (51%) |
| Age, Mean (SD) years |
38.4(8.1) |
37.0(10.1) |
31.6(8.2) |
| Range, years |
24-51 |
19-54 |
20-55 |
| BMI, [mean(SD) kg/m2] |
23.2(0) |
42.6(15.4) |
34.4(9.1) |
| Race |
| Caucasian |
24 |
25 |
34 |
| African-American |
1 |
1 |
3 |
| Other |
0 |
0 |
2 |
Table 1: Participant characteristics.
Global DNA Methylation
DNA extraction was performed on all peripheral blood specimens collected in EDTA vacutainer tubes using the DNeasy Blood & Tissue Kit obtained from Qiagen (Valencia, CA) following instructions provided by the manufacturer and described elsewhere [32]. Global DNA methylation was assessed using two different methodologies performed on the same DNA samples. The first method incorporated the use of the Human LINE-1 retrotransposable element commercially analyzed through EpigenDx (Worchester, MA), a company specializing in genomic methylation assays. EpigenDx performed the assay, validation, bisulfite conversion, PCR amplification and pyrosequencing to detect cytosine [C] to thymine [T] conversion from [uracil (U)] in the unmethylated (active) allele at CpG islands. The degree of methylation was analyzed as a ‘C/T SNP’ using AQ mode of computer analysis software program (https://epigendx.com/d/service/pyrosequencing/global-dna-methylation/). Additionally, four CpG island sites were studied using PCR primers which represent the DNA methylation status for select genes with methylation grouped accordingly (high, medium, or low) relative to lean controls, utilizing previously established protocols with detailed explanation described elsewhere [33]. The degree (amount) of unknown original target sequence was then compared with the sequence after bisulfite conversion [34].
The second approach for global methylation analysis was developed and commercially available through Epigentek (Farmingdale, NY) using a 5-methylcytosine based immunoassay platform (MethylFlashTM Global DNA Methylation () ELISA Kit, (https://www.epigentek.com/catalog/methylflash-global-dna-methylation-mc-elisa-easy-kitcolorimetric-p-5370.html). Purified sample DNA (100 ng) and unmethylated (negative) control DNA (10 ng) was incubated in strip wells with a specially developed solution to promote DNA binding and adherence to the sample well. Sample wells were treated with dilute 5 mC capture and detection antibodies to measure the methylated fraction of DNA which was quantified colorimetrically by absorbance readings using a microplate spectrophotometer. The percentage of methylated DNA was calculated as a proportion of the optical density (OD).

Methylation status was further analyzed for individual target genes known to be associated with obesity or with the causation of features seen in PWS. The complex SNRPN locus representing an important imprinted gene in the 15q11-q13 region plays a significant role in PWS and was examined using both the EpigenDx and Epigentek services. The SNRPN gene was used as a positive causative imprinted PWS gene control [6]. Other known imprinted genes, H19 and GNAS outside of chromosome 15 [35,36], along with a constituently active housekeeping gene, GAPDH [37], and a known obesity-related gene, BDNF [38] were tested and analyzed using the EpigenDx methodology and DNA methylation status of identified restriction sites quantified by PCR pyrosequencing using primers designed for LINE-1 elements (Table 2).
| Selected Loci |
| PWS imprinted mutation control |
Examination of "Cis/Trans" effects |
| SNRPN, 15q11.2 |
| Imprinting control genes |
|
| GNAS, 20q13.32 |
Co-localized on Chromosome 11 |
| H19, 11p 15.5 |
| Active gene (low methylation control) |
| BDNF,11p14.1 |
| GAPDH,12p13.31 |
|
Table 2: Epigen Dx target gene methylation analysis flowchart.
Other representative imprinted genes from the 15q11-q13 region (NDN, MAGEL2, MRKN3, IPW), non-imprinted 15q11- q13 genes (GABRB3, P) and the maternally expressed UBE3A gene were evaluated using the Methylation-Specific (MS)-PCR assay through the Epigentek laboratory services (https://www.epigentek.com/services/dna-methylation-analysis/methylationspecific-pcr/). Input DNA was subjected to bisulfite treatment according to standard protocols and MS-qPCR performed using the Methylamp MS-qPCR Kit with the following primers:
GABRB3 (117bps) #L04311
F : 5'-CGGCGGCGGTAGTAGTTAG-3'
R : 5'-AAAACCTTCCTCCCGCAA-3'
IPW (119 bps) #NG_021193
F: 5'-TTAACGTAGTTATTAGTTGG-3'
R: 5'-ACACGAAAATTACTAAACC-3'
MAGEL2 (120 bps) #NM_016776
F: 5'-TTTTATTCGTGATTCGTTAG-3'
R: 5'-GAATCAACGACGAAACCT-3'
MKRN3 (115 bps) #NM_005664
F: 5'-AATATAACGAAGCGTGTATGA-3'
R: 5'-CCTCTACTTCGCTATATTCC-3'
NDN (97 bps) #AB007828
F: 5'-TTTAGTCGTTGGTTAAGGTCG-3'
R: 5'-TTACTAACGCAACGCCTTC-3'
P (133 bps) #U19152
F: 5'-GTCGAGTGGGGAGTGTTGT-3'
R: 5'-CGCACCTCCGCTCTAAATA-3'
SNRPN (101 bps) #U41384
F: 5'-TCGTTGTAGTAGCGGTAGGT-3'
R: 5'-CCCTACACTACGACAAACAAA-3'
UBE3A (79 bps) #AH005553
F: 5'-CGGGGTGATTATAGGAGACG-3'
R: 5'-AATACGCGAACGAACGAAAC-3''
The Methylation Index (MI) was calculated for each gene target relative to β-actin using the following equation and water as a control:

Statistical Analysis
Summary data (% Methylation and MI) were received from both EpigenDx and Epigentek laboratory services and analyzed statistically using the Statistical Analyses System (SAS) software (version 9.4) (SAS Institute Inc, Cary, NC). Global and target sequence DNA methylation signal intensities were examined using analysis of variance (ANOVA) with Bonferroni correction to assess main and interactive effects of group, gender and target gene on DNA methylation readings from both LINE-1 and 5-methylcytosine based targeting immunoassay platforms.
Results
We utilized two different methods to evaluate the global DNA methylation patterns among individuals with nonsyndromic obesity, lean and PWS as well as gene methylation patterns at selected candidate obesity gene loci. Methylation status was assessed via LINE-1 elements within the genome at selected sites by EpigenDx and with the use of methylation detection by 5-methylcytosine based immunoassay methods of the CpG islands undertaken by Epigentek. Each service provided methylation levels from the same DNA samples.
LINE-1 methylation analysis
The results of the LINE-1 assay for global methylation relative to select target gene loci for SNRPN, imprinted (GNAS, H19) and low methylation (GAPDH, BDNF) active control genes are shown in Figure 1.
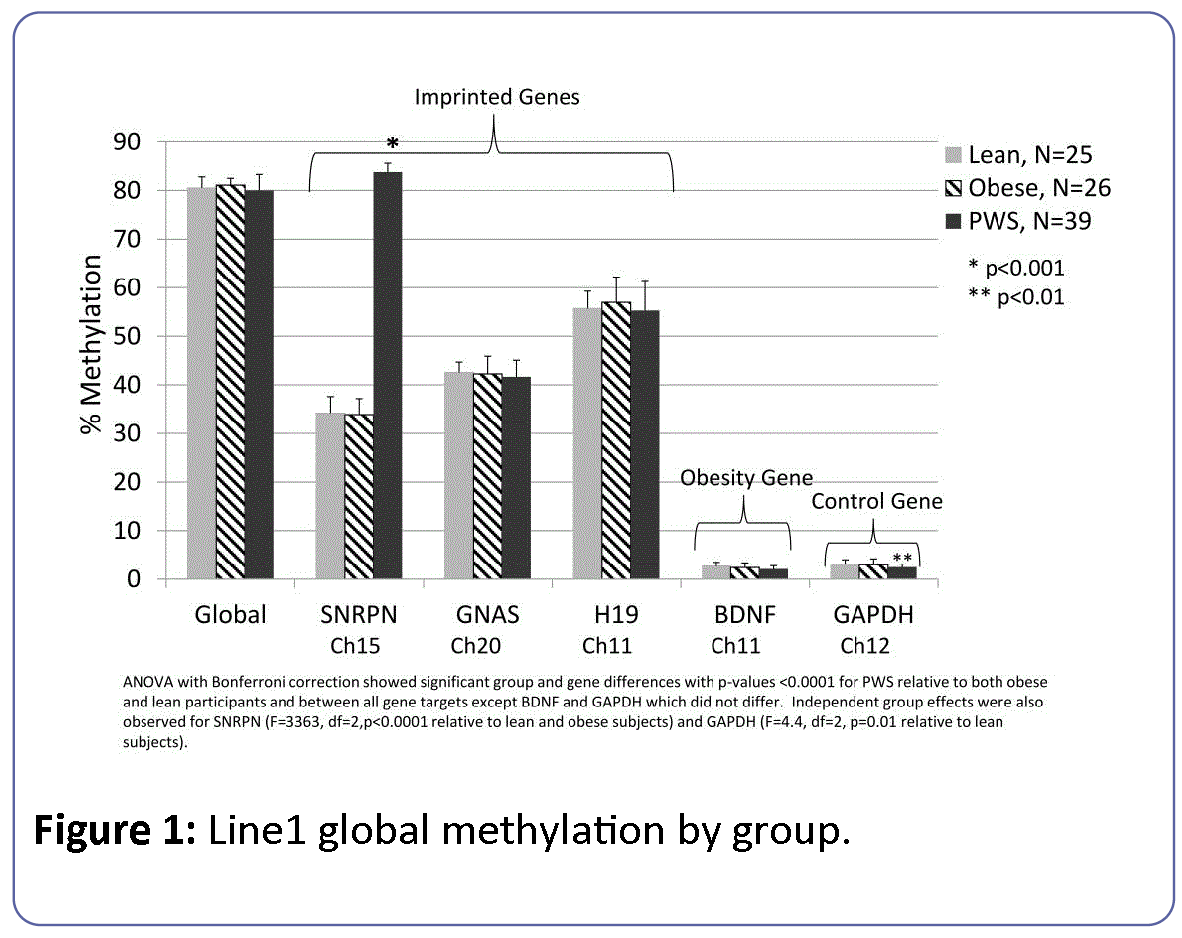
Figure 1: Line1 global methylation by group.
Global methylation levels were about 80% among all subject groups or about 1 fold higher than the imprinted target genes (GNAS, H19) and about 25 fold higher than the non-imprinted target genes (BDNF, GAPDH). The level of DNA methylation at GNAS and H19 were 40% and 50%, respectively and approximately 16 fold higher than the 3% methylation levels observed for BDNF and GAPDH loci independent of subject group or sex of participants. The methylation level for SNRPN was elevated at about 1.4 fold among PWS subjects relative to lean and obese participants. The overall regression model of methylation levels for global and target markers (SNRPN, GNAS, H19, GAPDH and BDNF) was significant (F=2880, df=18, p<0.0001). There was a significant difference by subject group (F=421, df=2, p<0.0001), target gene marker (F=9253, df=5, p<0.0001) and a subject group by marker interaction (F=473, df=10, p<0.0001) effect. However, there were no differences associated with participant gender (F=0.19, df=1, p=0.66). These results reflected the well-characterized effects of the imprinting error leading to PWS that produces significantly elevated methylation at the SNRPN locus among PWS subjects relative to both obese and lean participants. Methylation at the H19 locus was also significantly higher than at the GNAS locus but no evidence of trans effects on imprinting status were noted outside of the SNRPN locus. Subgroup analyses modeling the effects of group, sex and group*sex on individual target genes further supported the significantly elevated methylation levels for the imprinted SNRPN gene locus among PWS participants (F=3363, df=2, p<0.0001) with PWS participants showing 1.3 fold higher levels of methylation for SNRPN than lean and obese subjects. There were no significant differences between the three subject groups for the other two imprinted genes, H19 or GNAS. When comparing PWS genetic subtypes, PWS subjects having the UPD genetic subtype showed a slight but significantly increased methylation level (0.4 fold) compared to subjects with the deletion status at the H19 gene locus found on chromosome 11 (F=8.11, p=0.01).
5 m immunoassay, 5 mC calculation and global DNA quantification/MS-PCR target gene analysis
Global methylation levels determined by the Epigentek methodology were 2.2 ± 1.1% (0.06-5.6%) and did not differ by participant group (Figure 2). The statistical model of global methylation considering group, sex and age was not significant (F=1.3, df=6, dferorr=76, p<0.28) with no independent effects of age or gender.
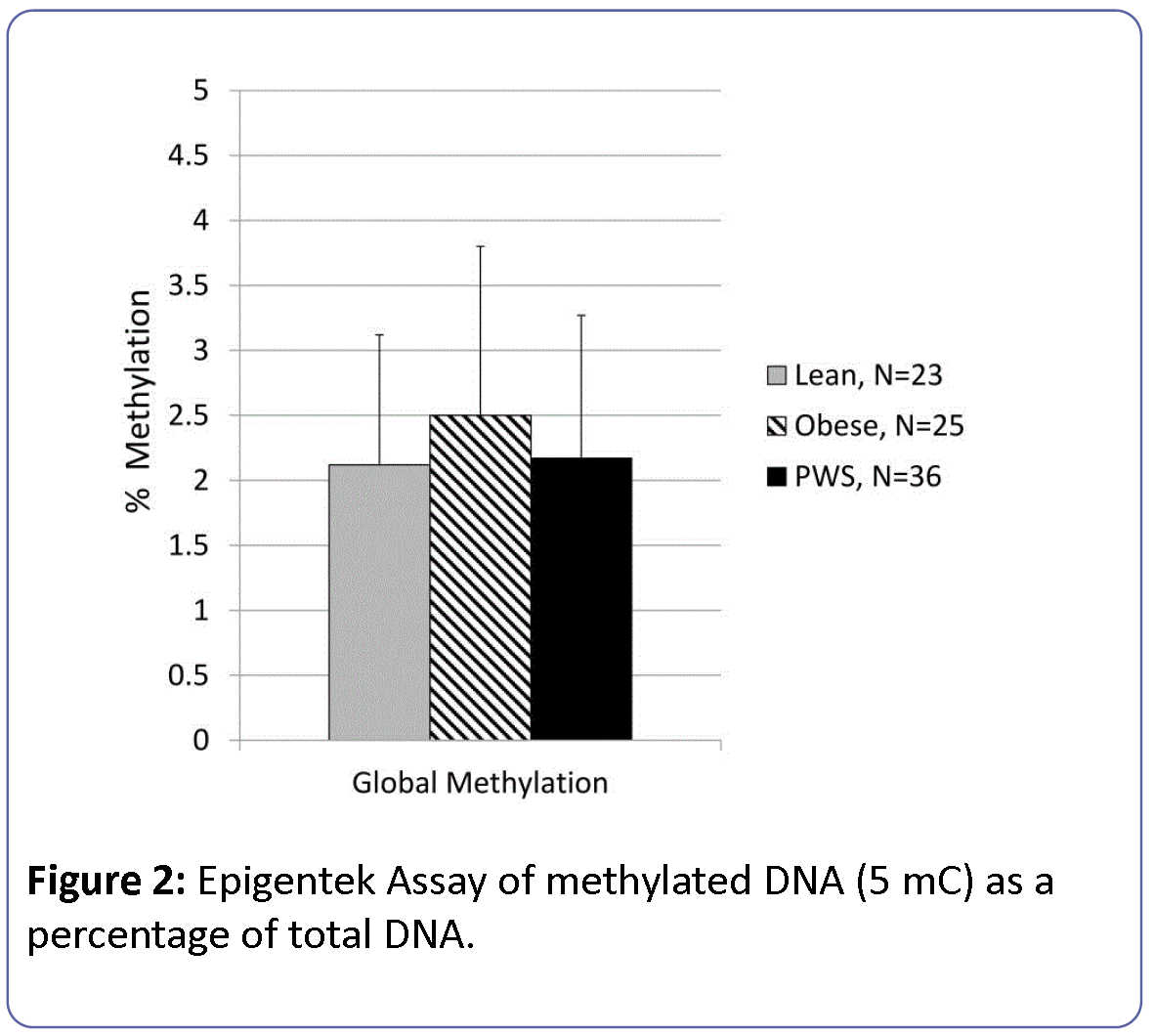
Figure 2: Epigentek Assay of methylated DNA (5 mC) as a percentage of total DNA.
Methylation levels for target genes measured by Epigentek were reported as a methylation index relative to β-actin and thus were not directly comparable to levels obtained from EpigenDx. PWS participants showed significantly greater overall methylation of paternally imprinted genes (SNRPN, IPW, NDN, MAGEL2, MKRN3) within the 15q11-q13 region than lean and obese participants. Methylation levels for NDN were significantly greater than SNRPN at the p=0.05 level of alpha for all groups ( Figure 3). ANOVA comparison of methylation for the imprinted genes (SNRPN, IPW, NDN, MAGEL2, MKRN3) from the 15q11-q13 region with Bonferroni correction controlling for group, gender and age was significant (F=14.8, df=16, p<0.0001) and showed significant differences between subject groups (F=17.8, df=2, p<0.0001) and imprinted target genes (F=41.8, df=4, p<0.0001), but no significant interaction effects between subject group and imprinted gene (F=1.6, df=8, p=0.13), gender (F=1.2, df=1, p=0.26) or age (F=0.13, df=1, p=0.72).
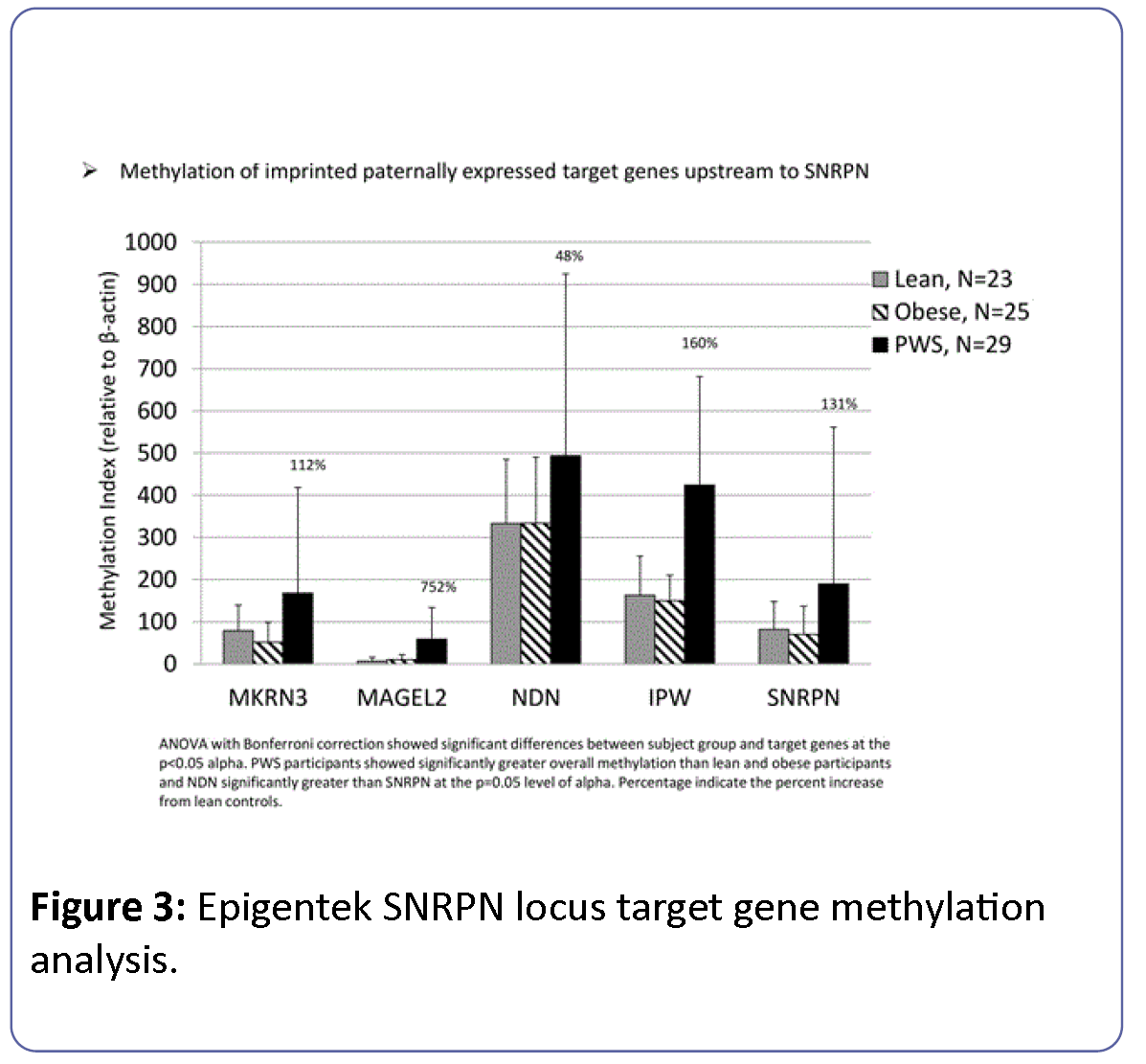
Figure 3: Epigentek SNRPN locus target gene methylation analysis.
Methylation of downstream target genes within the 15q11- q13 region (SNRPN, UBE3A, GABRB3, P) showed uniformly high levels of methylation at the UBE3A locus and significantly reduced methylation at the GABRB3 locus among all study groups (Figure 4). PWS participants showed significantly increased methylation at the SNRPN locus relative to lean and obese subject groups and significantly reduced methylation at the GABRB3 locus. ANOVA with Bonferroni correction controlling for gender and age was significant (F=19, df=13, p<0.0001) but showed no significant differences between subject groups (F=0.82, df=2, p<0.44). Differences between downstream target genes were statistically significant (F=70, df=3, p<0.0001) with a significant interaction between subject group and marker gene (F= 2.9, df=6, p=0.01). The effects of age and gender were not significant.
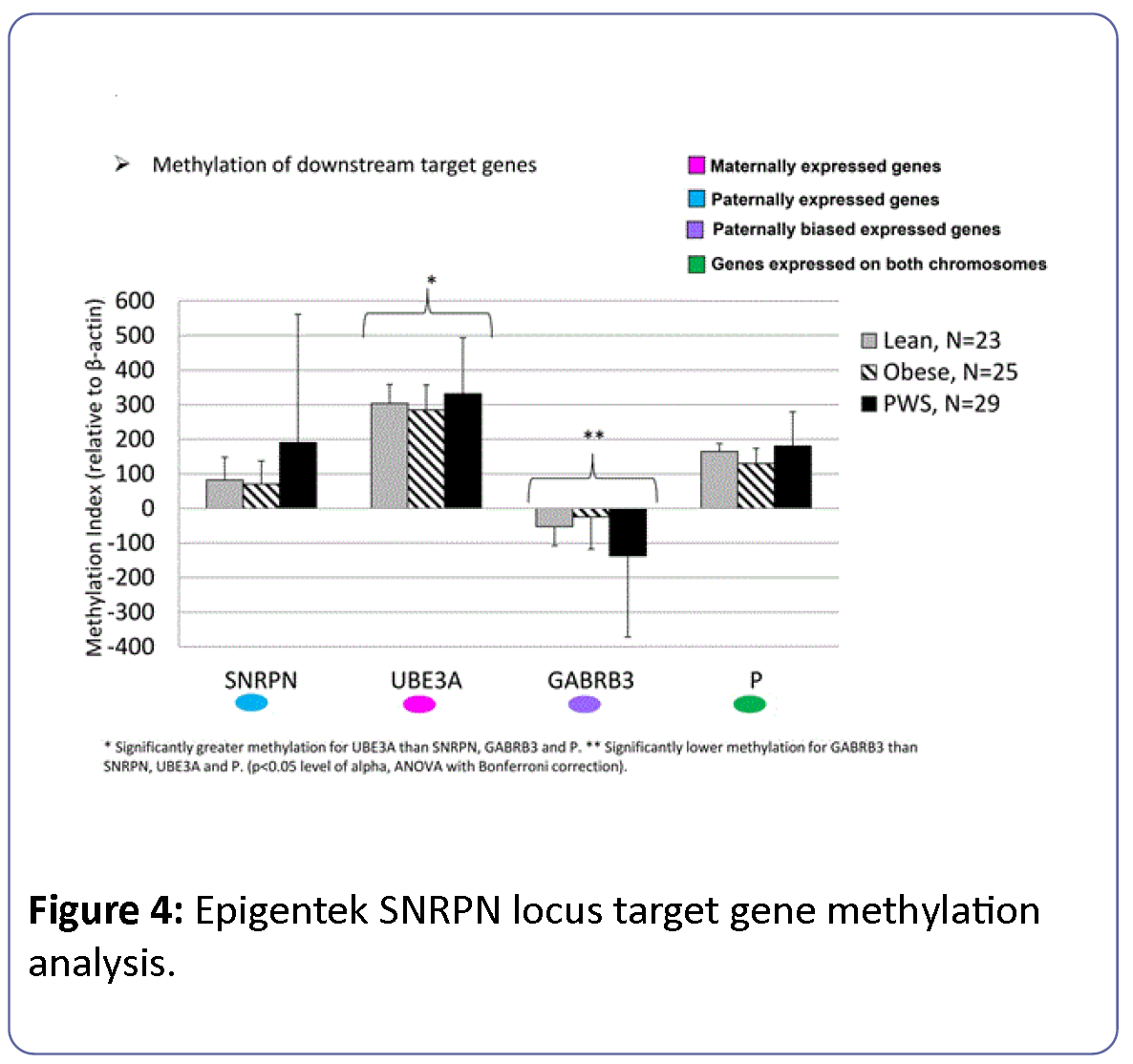
Figure 4: Epigentek SNRPN locus target gene methylation analysis.
The relationship between methylation index and PWS genetic subtype was modeled in PWS participants alone and considering the effects of target gene, gender and age. The overall model was significant (F=9.8, df=17, p<0.0001) and indicated a significant main effect of PWS genetic subtype (F=16.2, df=1, p<0.0001), gender (F=6.1, df=1, p<0.02) and target gene (F=20.4, df=7, p<0.0001) (Figure 5). Methylation index for PWS individuals with deletion subtype was consistently higher than for participants with the UPD15 genetic subtype. Furthermore, these effects appeared to select for paternally imprinted genes (e.g., MAGEL2, MKRN3) rather than maternally imprinted (e.g., UBE3A) or nonimprinted (e.g., P) genes.
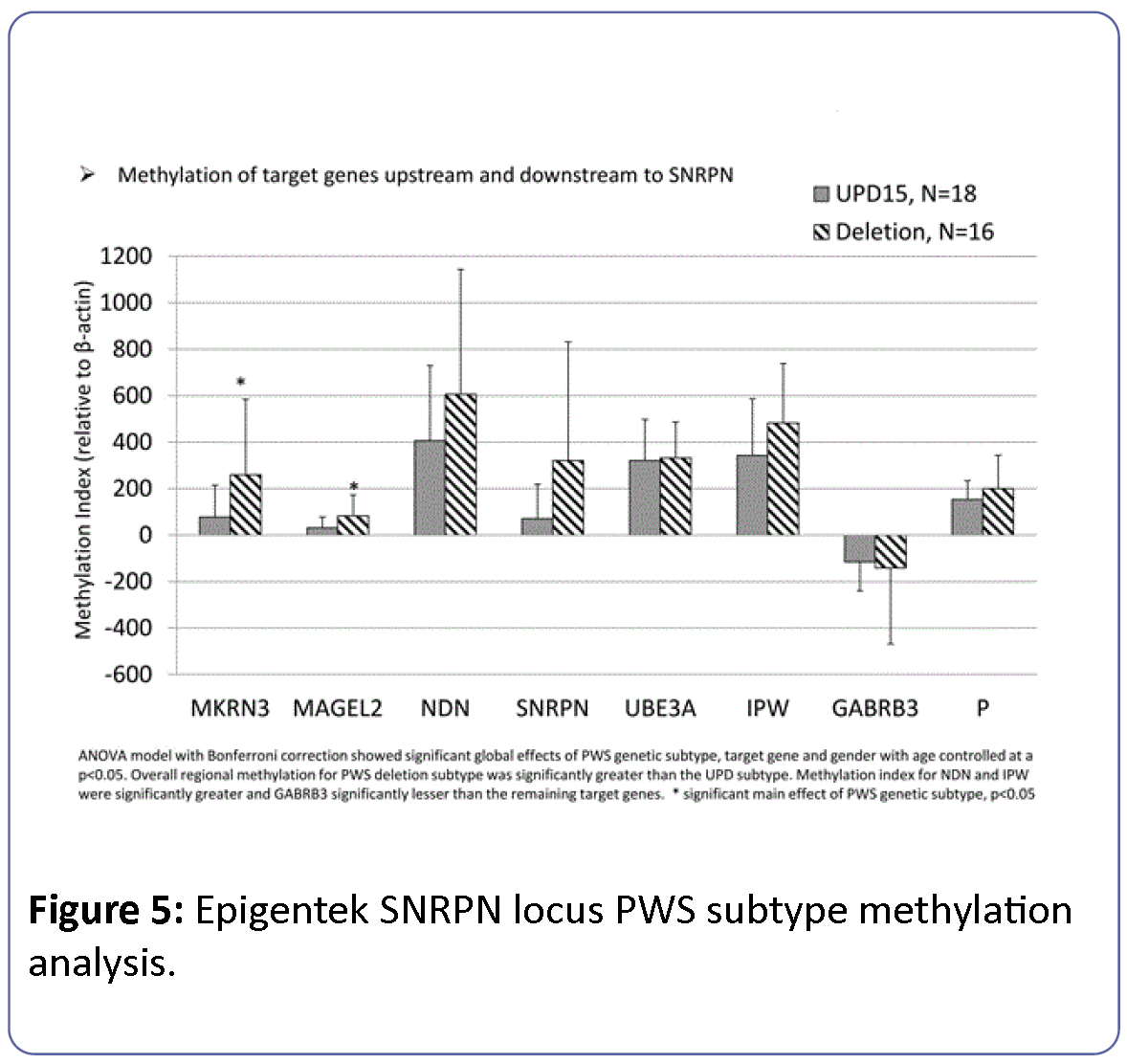
Figure 5: Epigentek SNRPN locus PWS subtype methylation analysis.
Discussion
The purpose of the current study was to analyze DNA methylation patterns among obese and lean individuals and individuals with PWS to test if obesity status is related to methylation levels, both globally and among selected single genes. We used two different methods through EpigenDx and Epigentek laboratory services to assess global methylation levels through the assay of two distinct genomic elements. The EpigenDx technology targeted methylation within LINE-1 repeat sequences which are known to be enriched in 5- methylated cytosine. Alternatively, Epigentek utilized an ELISA based immunoassay and antibodies sensitive to 5-methyl cytosine to detect all 5 mC distributed across the genome. Targeted examination of methylation at LINE-1 repeat sequences yielded higher global methylation levels (% methylation) than the more general antibody-mediated methodology - assessing all CpG islands in the genome. However, no differences in global methylation levels were observed between obese, lean and PWS participants using either methodology. Further, no significant disturbances were evident in the responses of obese individuals which correlated with measures for lean individuals in global and target gene assessments. There also were no significant differences in global methylation or target gene methylation between males and females. The relatively high methylation levels obtained from the LINE-1 assay were consistent with previous reports of methylation rates for this target sequence. The 5-mCytosine immunoassay probed a larger total number of CpG islands within the genome, and consequently, yielded lower overall methylation percentages which were also consistent with previous reports utilizing this technique. Neither methodology identified global methylation differences related to disease state or obesity status.
Both laboratory services were able to detect and confirm the methylation error at the SNRPN locus diagnostic of PWS. Further, selected positive and negative control genes of known imprinting status showed normal methylation profiles in PWS and obese individuals relative to lean. Genes that are imprinted are susceptible to environmental influences as these factors can alter gene expression and activity without changing the DNA molecule sequence [39]. Imprinted gene disturbances have been connected with human diseases including cancer, obesity, and neurological disorders with different aspect of behavior and language development [40,41,42], as well as genetic disorders such as Prader-Willi and Angleman syndromes. It has been estimated that more than 100 conditions result from disturbances in genetic imprinting. There is also evidence to suggest that assisted reproductive technology (ART) may alter imprinted gene regulation and increase chances of imprinting defects.
The GNAS gene located at chromosome 20q13.11 is a hormonally regulated, imprinted gene implicated in an obesity phenotype and syndrome (e.g., Albright hereditary osteodystrophy), and uses alternative promoters and splicing mechanisms to produce multiple transcripts which may be expressed biallelically or monoallelically from either the paternal or the maternal GNAS allele. The H19 gene located at chromosome 11p15.5 was the first imprinted non-coding RNA (ncRNA) transcript identified and abnormal methylation of H19 is associated with overgrowth and obesity [e.g., seen in Beckwith-Wiedemann syndrome (BWS)]. Disturbances in GNAS expression have been reported in individuals with BWS indicating a generalized methylation problem [43], involving the separate chromosome segments on different chromosomes with trans effects. Methylation status of these imprinted markers in our study appeared to be normal in PWS and obese individuals relative to lean showing no evidence of obesity-related disturbance or trans effects of the PWS genetic lesion.
Targeted examination of genes within the PWS critical 15q11-q13 genomic region confirmed an impact of the PWS genetic lesion on methylation of upstream and downstream effectors in PWS (so called cis effects), but the change in methylation level of nearby genes did not appear to be related to the proximity to the imprinting loci. As a maternally imprinted gene, UBE3A, showed the highest methylation index of downstream genes profiled. GABRB3 showed the lowest levels with significantly reduced methylation consistent with the paternally biased expression pattern. The PWS genetic subtype also appears to differentially impact gene expression in the region with significantly greater methylation observed for the imprinted target genes among deletion vs. UPD15 PWS subtypes. There are several possible hypothetical mechanisms which may be proposed to account for this observation. A chromosome deletion and associated copy number change may produce an increased proportional methylation response (per total input DNA) relative to the balanced copy number in UPD15 from the contributions of two maternal alleles. However, the selection for imprinted genes and those with paternally biased gene expression is not consistent with a copy-number related imbalance theoretically impacting the entire region. It is also possible that the methylation signal in UPD15 engages feedback regulatory mechanisms leading to relaxation of the imprint in neighboring genes - partially normalizing methylation in the region. Alternatively, the deletion mutation may engage regulatory mechanisms that promote methylation of the region as a means to mitigate the impact of the genetic error through processes analogous to the inactivation of fragile X protein as an example. Such a subtype-mediated variation in regional genomic imprinting in PWS could further complicate assessments and add variation to the observed PWS phenotype.
Conclusion
The purpose of the current study was to further advance our understanding of the connection between genetics and obesity by analyzing differences and similarities in global and gene methylation patterns in known imprinted syndromes (i.e., PWS) or in disease states (i.e., obesity) and lean individuals. Hence, we compared methylation patterns of obese and lean individuals with those diagnosed with PWS. Our results showed no global methylation change among nonsyndromic obese and PWS individuals, as compared to lean controls using technology described in our study. Our analysis also provided a more detailed characterization of the imprinting status in the critical 15q11-q13 PWS genomic region and a framework to gauge the regional impact of the PWS genetic lesion on neighboring genes and we confirmed an impact on upstream and downstream effectors in PWS. The results of our analysis may provide the framework for larger studies for an expanded characterization of methylation disturbances in those with PWS and obesity and further examine methylation and PWS genetic subtypes as well as the influence of obesity level, age and gender on methylation status. Broader assessments should examine the interrelationship between DNA methylation, obesity status and hormone levels (e.g., estrogen) in the hope of developing novel treatment strategies and interventions to improve quality of life for PWS individuals and possibly reduce obesity rates in the general population as related to gene interaction and methylation status impacting gene function.
Acknowledgments
We would like to acknowledge Austen McGuire for his assistance in manuscript preparation. We acknowledge financial support from the Institute for Reproductive Health and Regenerative Medicine (IRHRM)-Pilot Project; the Prader- Willi Syndrome Association (USA) and NICHD grant 025258. Further we would like to thank PWS patients and families for their ongoing support and participation in PWS research.
Conflicts of Interest
The authors declare no conflicts of interest.
Statement of Ethics
The study protocol was approved and monitored by the University of Kansas Medical Center Institutional Review Board for research involving human subjects and all subjects (or their parents or guardians) have given their informed written consent to participate.
References
- Kelly T, Yang W, Chen CS, Reynolds K, He J (2008) Global burden of obesity in 2005 and projections to 2030. Int J Obes (Lond) 32: 1431-1437.
- Bear MF (2005) Therapeutic implications of the mGluR theory of fragile X mental retardation. Genes Brain Behav 4: 393-398.
- Butler MG (2011) Prader-Willi syndrome: Obesity due to genomic imprinting. Curr Genomics 12: 204-215.
- Marshall JD, Beck S, Maffei P, Naggert JK (2007) Alström syndrome. Eur J Hum Genet 15: 1193-1202.
- Butler MG, Wang K, Marshall JD, Naggert JK, Rethmeyer JA, et al. (2015) Coding and noncoding expression patterns associated with rare obesity-related disorders: Prader-Willi and Alström syndromes. Adv Genomics Genet 2015: 53-75.
- Bittel DC, Butler MG (2005) Prader-Willi syndrome: clinical genetics, cytogenetics and molecular biology. Expert Rev Mol Med 7: 1-20.
- Butler MG, Hanchett JM, Thompson T (2006) Clinical findings and natural history of PWS. In: Butler MG, Lee PDK, Whitman BY, eds. Management of Prader-Willi Syndrome. 3rd ed. New York, NY: Springer Science and Business Media 1-48.
- Cassidy SB, Schwartz S, Miller JL (2012) Prader-Willi syndrome. Genet Med 14: 10-26.
- Angulo MA, Butler MG, Cataletto ME (2015) Prader-Willi syndrome: A review of clinical, genetic, and endocrine findings. J Endocrinol Invest 38: 1249-1263.
- Moore LD, Le T, Fan G (2013) DNA methylation and its basic function. Neuropsychopharmacology 38: 23-38.
- Haig D, Graham C (1991) Genomic imprinting and the strange case of the insulin-like growth factor II receptor. Cell 64: 1045-1046.
- Butler MG (2009) Genomic imprinting disorders in humans: a mini-review. J Assist Reprod Genet 26: 477-486.
- Platonov ES, Isaev DA (2006) Genomic imprinting in the epigenetics of mammals. Genetika 42: 1235-1249.
- Surani MA, Barton SC, Norris ML (1984) Development of reconstituted mouse eggs suggests imprinting of the genome during gametogenesis. Nature 308: 548-550.
- Delaval K, Wagschal A, Feil R (2006) Epigenetic deregulation of imprinting in congenital diseases of aberrant growth. Bioessays 28: 453-459.
- Luedi PP, Hartemink AJ, Jirtle RL (2005) Genome-wide prediction of imprinted murine genes. Genome Res 15: 875-884.
- Zakharova IS, Shevchenko AI, Zakian SM (2009) Monoallelic gene expression in mammals. Chromosoma 118: 279-90.
- Hoffmann MJ, Schulz WA (2005) Causes and consequences of DNA hypomethylation in human cancer. Biochem Cell Biol 83: 296-321.
- Kramerov DA, Vassetzky NS (2005) Short retroposons in eukaryotic genomes. Int Rev Cytol 247: 165-221.
- Sellis D, Provata A, Almirantis Y (2007) Alu and LINE1 distributions in the human chromosomes: evidence of global genomic organization expressed in the form of power laws. Mol Biol Evol 24: 2385-2399.
- Chueh AC, Northrop EL, Moore KH, Choo KH, Wong LH (2009) LINE retrotransposon RNA is an essential structural and functional epigenetic component of a core neocentromeric chromatin. PLoS Genet 5: 1000354.
- Doucet AJ, Hulme AE, Sahinovic E, Kulpa DA, Moldovan JB, et al. (2010) Characterization of LINE-1 ribonucleoprotein particles. PLoS 6:1001150.
- Kazazian HH (2004) Mobile elements: Drivers of genome evolution. Science 303: 1626-1632.
- Weisenberger DJ, Campan M, Long TI, Kim M, Woods C, et al. (2005) Analysis of repetitive element DNA methylation by MethyLight. Nucleic Acids Res 33: 6823-6836.
- Han JS, Boeke JD (2005) LINE-1 retrotransposons: Modulators of quantity and quality of mammalian gene expression? Bioessays 27: 775-784.
- Häsler J, Strub K (2006) Alu elements as regulators of gene expression. Nucleic Acids Res 34:5491-5497.
- Garcia-Lacarte M, Milagro FI, Zulet MA, Martinez JA, Mansego ML (2016) LINE-1 methylation levels, a biomarker of weight loss in obese subjects, are influenced by dietary antioxidant capacity. Redox Rep 21: 67-74.
- Godfrey KM, Sheppard A, Gluckman PD, Lillycrop KA, Burdge GC et al. (2011) Epigenetic gene promoter methylation at birth is associated with child’s later adiposity. Diabetes 60: 1528-1534.
- Perng W, Mora-Plazas M, Marín C, Rozek LS, Baylin A (2013) A prospective study of LINE-1DNA methylation and development of adiposity in school-age children. PLoS One 8: 62587.
- Na YK, Hong HS, Lee DH, Kim DS (2014) Effect of body mass index on global DNA methylation in healthy Korean women. Mol Cells 37: 467-472.
- Butler MG (1990) Prader-Willi syndrome: current understanding of cause and diagnosis. Am J Med Genet 35: 319-32.
- Rethmeyer JA, Tan X, Manzardo A, Schroeder SR, Butler MG (2013) Comparison of biological specimens and DNA collection methods for PCR amplification and microarray analysis. Clin Chem Lab Med 51: 79-83.
- Yang I, Park IY, Jang SM, Shi LH, Ku HK, et al. (2006) Rapid quantification of DNA methylation through dNMP analysis following bisulfite-PCR. Nucleic Acids Res 34: 61.
- Liao LM, Brennan P, van Bemmel DM, Zaridze D, Matveev V, et al. (2011) LINE-1 methylation levels in leukocyte DNA and risk of renal cell cancer. PLoS One 6: 27361.
- Bastepe M, Juppner H (2005) GNAS locus and pseudohypoparathyroidism. Horm Res 63: 65-74.
- Reik W, Walter J (2001) Genomic imprinting: parental influence on the genome. Nat Rev Genet 2: 21-32.
- Barber RD, Harmer DW, Coleman RA, Clark BJ (2005) GAPDH as a housekeeping gene: Analysis of GAPDH mRNA expression in a panel of 72 human tissues. Physiol Genomics 21: 389-395.
- Butler MG, McGuire A, Manzardo AM (2015) Clinically relevant known and candidate genes for obesity and their overlap with human infertility and reproduction. J Assist Reprod Genet 32: 495-508.
- Jirtle RL, Skinner MK (2007) Environmental epigenomics and disease susceptibility. Nat Rev Genet 8: 253-262.
- Bartolomei MS, Tilghman SM (1997) Genomic imprinting in mammals. Annu Rev Genet 31: 493-525.
- Murphy SK, Jirtle RL (2003) Imprinting evolution and the price of silence. Bioessays 25: 577-588.
- Walter J, Paulsen M (2003) Imprinting and disease. Semin Cell Dev Biol 14: 101-10.
- Bliek J, Verde G, Callaway J, Maas SM, De Crescenzo A, et al. (2009) Hypomethylation at multiple maternally methylated imprinted regions including PLAGL1 and GNAS loci in Beckwith-Wiedemann syndrome. Eur J Hum Genet 17: 611-6199.

