Simonetta Friso1*, Francesca Pizzolo1, Silvia Udali1, Patrizia Guarini1, Annalisa Castagna1, Letizia Consoli1, Gianluca Salvagno2, Elisa Tinazzi1, Patrizia Pattini1, Sang-Woon Choi3,4, Claudio Lunardi1 and Oliviero Olivieri1
1Department of Medicine, University of Verona, School of Medicine, Piazzale L.A. Scuro 10, 37134 Verona, Italy
2Department of Pathology and Diagnostics, University of Verona, School of Medicine, Piazzale L.A. Scuro 10, 37134 Verona, Italy
3Chaum Life Center, CHA University, 442 Dosan-daero, Gangnam-gu, Seoul, 135-948, Korea
4Tufts University School of Nutrition Science and Policy, 150 Harrison Ave, Boston, MA 02111, USA
*Corresponding Author:
Simonetta Friso
University of Verona, School of Medicine, Department of Medicine
Policlinico “G.B. Rossi”, P.le L.A. Scuro 10, 37134 Verona, Italy
Tel: +39-045-8126490
Fax: +39-045-580111
E-mail: simonetta.friso@univr.it
Received date: December 24, 2015; Accepted date: January 22, 2016; Published date: January 30, 2016
Citation: Friso S, Pizzolo F, Udali S, et al. Epigenetic Regulation of HSD11B2 Gene by Promoter Methylation in Glucocorticoid- Treated Patients. J Clin Epigenet. 2016, 2:1. DOI: 10.21767/2472-1158.100012
Copyright: © 2016 Friso S, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Keywords
HSD11B2 ; Hypertension; Cortisone; Epigenetics; DNA methylation
Abbreviations
11 beta-HSD2: 11 Beta-Hydroxysteroid Dehydrogenase 2; THFs/THE: Tetrahydrocortisol/Tetrahydrocortisone; PBMCs: Peripheral Blood Mononuclear Cells; THF: Tetrahydrocortisol; αTHF: 5α-Tetrahydrocortisol; THE: Tetrahydrocortisone
Introduction
The function of 11 beta-hydroxysteroid dehydrogenase type 2 (11 beta-HSD2) (EC 1.1.146) has a prominent role in essential hypertension [1-3] where a loss of its activity leads towards the activation of mineralocorticoid receptors by cortisol causing renal sodium retention [4] and high blood pressure [5,6]. The HSD11B2 mRNA expression, furthermore, has been shown to be inversely related to urinary tetrahydrocortisol (THFs)/ tetrahydrocortisone (THE) ratio [7], evidence that the THFs/ THE ratio depends on the 11beta-HSD2 activity [8,9] and may, therefore, represent a useful surrogate biomarker of the 11beta- HSD2 enzyme function [10]. Interestingly, promoter methylation at HSD11B2 site was demonstrated to be a functional mechanism for the transcriptional regulation of this gene and, thus, a reliable indicator of 11beta-HSD2 enzyme activity [11].
In a previous study [12] we observed that glucocorticoid-treated patients developing arterial hypertension showed a higher HSD11B2 promoter methylation concomitant with a higher urinary THFs/THE ratio. Furthermore, essential hypertensive patients with elevated urinary THFs/THE ratio showed higher HSD11B2 promoter methylation [12]. It is well-established that epigenetic mechanisms are potentially reversible, whereas the hypothesis that cortisol might transiently modulate HSD11B2 promoter methylation and eventually influence urinary THFs/ THE ratio and the onset of hypertension needs to be confirmed. In fact this mechanism could be proven by assessing the gene promoter methylation and the urinary THFs/THE ratio before and during steroid therapy and after therapy withdrawal.
Precisely to address this hypothesis and with the aim of unraveling a possible role of epigenetic phenomena as the potential underlying mechanism in the pathogenesis of steroid-therapy induced arterial hypertension, we designed the present study. Patients affected by rheumatologic diseases requiring the usage of prednisone for the acute phase of disease, were evaluated for the HSD11B2 promoter methylation status as well as for urinary THFs/THE ratio before, during and after withdrawal of prednisone therapy.
Materials and Method
Study design
The study protocol conformed to the ethical guidelines of the 1975 Declaration of Helsinki and was approved by the Ethical Review Board of the University of Verona School of Medicine Hospital (Verona, Italy). Written informed consent was obtained from each patient after a detailed explanation of the study. Six subjects were enrolled among those referring to the Unit of Clinical Immunology of the Verona University Hospital (Verona, Italy) and written informed consent was obtained from each patient after a detailed explanation of the study. Patients were four males and two females (age range 34-60 years), prescribed to undergo prednisone therapy for autoimmune diseases in active phase (four sarcoidosis, one autoimmune eosinophilia and one autoimmune neurosensorial hypoacusia). All patients ingested 75 mg of prednisone daily (about 1 mg/kg per day), except the patient affected by autoimmune neurosensorial hypoacusia (37.5 mg per day, 0.5 mg/kg per day), for a period of time of one month.After the remission of the disease verified by clinical and biochemical analysis, patients started a prednisone slow décalage regimen up to the complete withdrawal of glucocorticoid therapy. The molecular and biochemical parameters were evaluated at enrolment (T0), at one month of therapy (T1) and at more than one year of complete withdrawal of steroid therapy (T2). No one of the patient was affected by hypertension nor was taking any other drug before and during therapy and after prednisone therapy suspension.
Biochemical analyses and urinary steroid metabolites
Venous blood samples were collected into EDTA-containing Vacutainer® tubes after overnight fasting for routine analysis including plasma creatinine and serum/urinary electrolytes. Cortisol was measured by a chemiluminescent enzyme immunoassay (Immulite® 2000 Cortisol, Siemens Medical Solutions Diagnostics, Los Angeles, CA, USA).
Samples for aldosterone and renin assay were collected midmorning (09:00–10:00 h) and the patient was maintained at least 30 min in the upright posture and 10 min in the seated position. Renin was measured as direct active renin by the LIAISON® Direct Renin assay and aldosterone by RIA (Radio Immuno Assay) (both by Sorin Biomedical Diagnostics, Vercelli, Italy) [13].
Urinary tetrahydrocortisol (THF), 5α-tetrahydrocortisol (αTHF) and tetrahydrocortisone (THE) were analyzed by gas chromatography/mass spectrometry [14,15].
DNA methylation analysis by bisulfite pyrosequencing
DNA extraction from peripheral blood mononuclear cells (PBMCs): Blood samples were drawn in EDTA-containing BD Vacutainer® tubes, centrifuged at 2,500 x g for 15 min at 4°C and the PBMCs collected. DNA was extracted from PBMCs samples with a standard phenol/chloroform procedure and DNA concentration and purity were assessed by NanoDrop 1000 spectrophotometer (Thermo Fisher Scientific, Wilmington, DE) and DNA was stored at -20°C.
HSD11B2 gene promoter bisulfite pyrosequencing: Pyrosequencing analysis was performed on two HSD11B2 promoter regions, namely Region 1 (bases -419 to -177, relative to the start of transcription) and Region 2 (bases -692 to -595, relative to the start of transcription). Genomic DNA (20 ng) was modified by sodium bisulfite using the EZ DNA Methylation kit (ZYMO Research, CA, USA) according to the manufacturer’s instructions.
The Region 1 was amplified using a couple of primers (Table 1) designed by PSQ Assay Design (Biotage AB, Uppsala, Sweden); to avoid the possible influence of methylation status on the amplification reaction, primers were designed in CpG-free regions. The Region 2 was amplified with previously reported primers [16] (Table 1).
| HSD11B2 promoter |
Product size |
Primer sequences (5’ ® 3’) |
PCR temperature profiles |
| Region 1 |
243 bp |
F: GGTGGTGAGATTAGTAAAGGGTAT
R: biotin- AACCCAAACAAAATCCCAAAATTACTAC
Seq: GGGAGTGTGGGTGGGGG |
94°C
10 min |
94°C 30 sec
56°C 30 sec
72°C 30 sec |
X 45 cycles |
72°C
10 min |
| Region 2 |
97 bp |
F: AAGTTTTGGAAGGAAAGGGAAAGA
R: biotin-ACAAAACCTACCTAAAACAAAAACTA
Seq: GGGGTAGAGATTTTAAGAA |
94°C
10 min |
94°C 30 sec
58°C 30 sec
72°C 30 sec |
X 45 cycles |
72°C
10 min |
F: forward primer, R: reverse primer, Seq: sequencing primer
Table 1. Primers and PCR temperature profiles for HSD11B2 pyrosequencing reaction.
The amplification reactions were performed in a 25 μL reaction volume with the primer sets and 5 Units of Taq polymerase (Solgent Co., Daejeon, Korea). The PCR conditions are detailed in Table 1. PCR products were visualized on a 1.5% agarose gel by ethidium bromide staining for verification. Pyrosequencing reactions were performed with the sequencing primers on the PSQ HS 96A System (Biotage AB, Sweden) according to the manufacturer’s specifications (Table 1). For each region, the methylation status of 4 CpGs sites was evaluated and expressed as percentage of methylation, mCyt/(mCyt+Cyt). The average DNA methylation (%) was then calculated for each region at HSD11B2 promoter site.
Statistical analysis
Distribution of continuous variables is expressed as mean ± SD. The correlation between variables was evaluated using Pearson’s correlation coefficient. The analysis of differences between mean values at T0, T1, and T2 time points was undertaken by non-parametric Wilcoxon test for paired samples. Repeated measures were further investigated using linear quantile mixedeffect models [17]. The two approaches yielded comparable results. Values of P<0.05 were considered statistically significant. Statistical analysis was performed using the IBM SPSS 20 statistical software (IBM Inc, Amonk, NY, USA) and R 3.1.3 (R Foundation for Statistical Computing, Vienna) with the lqmm package [17].
Results
Table 2 shows the clinical and biochemical characteristics of the patients at the three time points: baseline (T0), following onemonth prednisone therapy (T1) , and at least one year after prednisone withdrawal (T2). As shown, systolic blood pressure slightly increased at T1 and decreased at T2 and diastolic blood pressure showed a mild increase at T1, but these differences did not reach the statistical significance. The THFs/THE ratio significantly increasedafter prednisone therapy (T0: 1.29 ± 0.80, T1: 4.10 ± 1.62, P=0.043), and decreased after therapy withdrawal, although not significantly (T1: 4.10 ± 1.62, T2: 1.09 ± 0.36, P=0.068) (Table 2, Figure 1). Among the biochemical parameters, serum aldosterone increased after therapy as compared to baseline (P=0.028).
| |
T0 |
T1 |
T2 |
P* |
P† |
| SBP (mm Hg) |
127 ± 6.1 |
131 ± 5.8 |
123 ± 5.0 |
N.S |
N.S |
| DBP (mm Hg) |
79 ± 5.8 |
84 ± 3.8 |
76 ± 11.1 |
N.S |
N.S |
| S-Na+ (mmol/L) |
140 ± 5.3 |
141 ± 3.0 |
141 ± 2.2 |
N.S |
N.S |
| S-K+ (mmol/L) |
3.85 ± 0.39 |
3.88 ± 0.41 |
3.73 ± 0.44 |
N.S |
N.S |
| S-Cl- (mmol/L) |
104 ± 2.3 |
104 ± 3.1 |
104 ± 1.3 |
N.S |
N.S |
| U-Na+ (mmol/24 h) |
77.3 ± 20.1 |
97.2 ± 54.2 |
70.0 ± 20.6 |
N.S |
N.S |
| U-K+ (mmol/24 h) |
59.0 ± 16.4 |
46.0 ± 10.7 |
40.7 ± 14.6 |
N.S |
N.S |
| U-Cl- (mmol/24 h) |
98.7 ± 54.5 |
104.3 ± 65.7 |
78.5 ± 28.1 |
N.S |
N.S |
| Renin (pg/mL) |
11.5 ± 15.4 |
21.8 ± 14.2 |
8.21 ± 4.47 |
N.S |
N.S |
| Aldosterone (pg/mL) |
165 ± 29.2 |
232 ± 65.4 |
227 ± 81.4 |
0.028 |
N.S |
| ARR |
36.1 ± 28.2 |
20.7 ± 21.5 |
36.8 ± 27.3 |
N.S |
N.S |
| THF (mg/24 h) |
2.43 ± 1.28 |
0.43 ± 0.19 |
5.52 ± 8.14 |
0.028 |
N.S |
| α-THF (mg/24 h) |
2.33 ± 2.15 |
0.26 ± 0.27 |
2.05 ± 2.46 |
0.028 |
N.S |
| THE (mg/24 h) |
3.09 ± 2.37 |
0.18 ± 0.09 |
6.46 ± 8.73 |
0.028 |
N.S |
| THFs/THE ratio |
1.29 ± 0.80 |
4.10 ± 1.62 |
1.09 ± 0.36 |
0.043 |
N.S |
Data are means ± SD. P-values refer to paired samples non-parametric Wilcoxon test for the comparison between T0 and T1 (*) or for the comparison between T1 and T2 (†).
T0, baseline; T1, after-prednisone therapy; T2, after-prednisone withdrawal; SBP, systolic blood pressure; DBP, diastolic blood pressure; ARR, aldosterone to renin ratio; THF, urinary tetrahydrocortisol; α-THF, 5α-tetrahydrocortisol; THE, tetrahydrocortisone; THFs/THE ratio, tetrahydrocortisolversus tetrahydrocortisone-metabolites ratio.
Table 2 Clinical and biochemical features of glucocorticoid-treated patients.
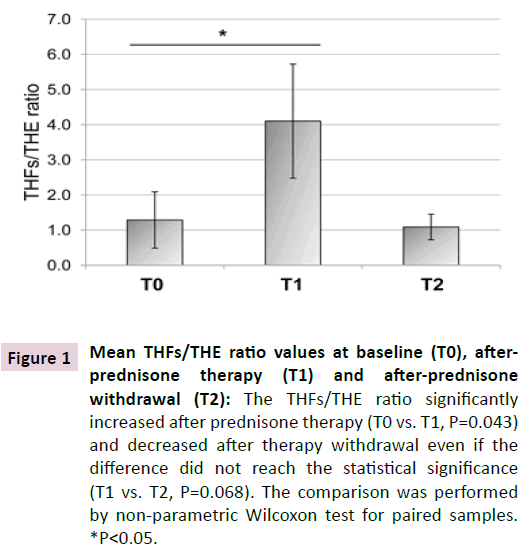
Figure 1 Mean THFs/THE ratio values at baseline (T0), afterprednisone therapy (T1) and after-prednisone withdrawal (T2): The THFs/THE ratio significantly increased after prednisone therapy (T0 vs. T1, P=0.043) and decreased after therapy withdrawal even if the difference did not reach the statistical significance (T1 vs. T2, P=0.068). The comparison was performed by non-parametric Wilcoxon test for paired samples. *P<0.05.
As for the pyrosequencing analysis of HSD11B2 gene promoter, Figure 2 reports the mean DNA methylation levels (%) at promoter Region 1 and Region 2. Overall, the two regions showed a different methylation status with lower levels associated to Region 1.
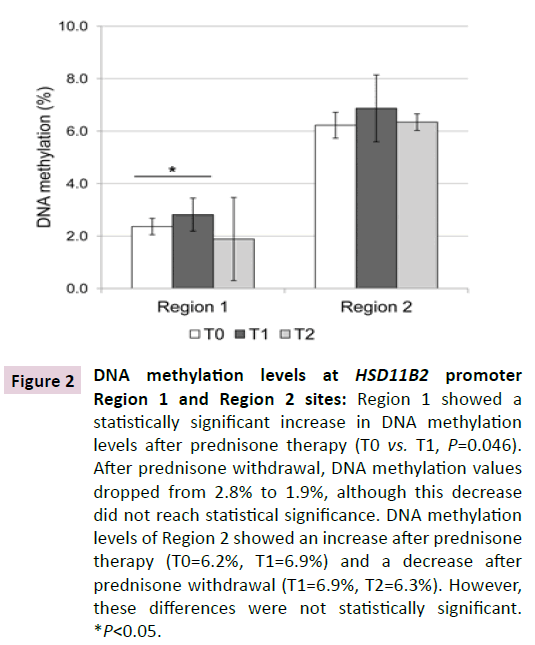
Figure 2 DNA methylation levels at HSD11B2 promoter Region 1 and Region 2 sites: Region 1 showed a statistically significant increase in DNA methylation levels after prednisone therapy (T0 vs. T1, P=0.046). After prednisone withdrawal, DNA methylation values dropped from 2.8% to 1.9%, although this decrease did not reach statistical significance. DNA methylation levels of Region 2 showed an increase after prednisone therapy (T0=6.2%, T1=6.9%) and a decrease after prednisone withdrawal (T1=6.9%, T2=6.3%). However, these differences were not statistically significant. *P<0.05.
After prednisone therapy (T1), Region 1 showed a statistically significant increase in DNA methylation levels (T0: 2.4%, T1 2.8%, P=0.046). After prednisone withdrawal (T2), DNA methylation values dropped from 2.8% to 1.9%, although this decrease did not reach the statistical significance.
At Region 2 the DNA methylation status variation showed a trend similar to Region 1, with an increase after prednisone therapy (from 6.2% to 6.9%) and a decrease after prednisone withdrawal (from 6.9% to 6.3%).
A positive correlation was observed between HSD11B2 promoter methylation at Region 2 and the THFs/THE ratio (Pearson’s correlation coefficient=0.537, P=0.039).
Discussion
This study shows that HSD11B2 promoter methylation increased after glucocorticoid therapy and decreased after its withdrawal in PBMCs DNA of patients taking prednisone at high-dose for one month. These changes clearly paralleled the increase and decrease of THFs/THE ratio, a well-recognized surrogate biochemical marker of the 11beta-HSD2 enzyme function. Compared to previous studies that used methyl-specific PCR, bisulfite pyrosequencing can more precisely measure the methylation status of each CpG site and therefore substantiate previous findings [12].
The higher methylation at HSD11B2 promoter following prednisone treatment and its trend to decrease after therapy interruption with a concomitant increase and subsequent decrease of THFs/THE ratio are consistent with the well-described functional role of promoter methylation [18] in the transcriptional repression of HSD11B2 [11,12] and ultimately in the regulation of enzyme activity.
A trend towards the increase of blood pressure following prednisone therapy was also documented, although the difference did not reach statistical significance. This result can be explained by the relatively short period of steroid treatment in patients that were normotensive at baseline. The THFs/THE ratio and HSD11B2 promoter methylation seems more sensitive to steroid therapy than the modification of blood pressure.
While these findings substantiate our previous study [12], they suggest some interesting considerations. They confirm the hypothesis that the altered function of 11beta-HSD2 may represent one of the possible mechanisms underlying the pathogenesis of hypertension during steroid therapy. Moreover, they highlight the involvement of an epigeneticallydriven gene regulation mechanism by methylation at HSD11B2 promoter site. Although it is well-established that epigenetic phenomena are potentially reversible, the present observation of their modulation in patients undergoing steroid therapy, as it specifically refers to HSD11B2 promoter methylation is an interesting element of novelty. Moreover, the demonstration that HSD11B2 promoter methylation is modifiable and leads to enzyme activity modulation as proven by changes observed in the THFs/THE ratio, may explain the described reversibility of hypertension developing under steroid therapy regimen.
A key point for discussion relies on steroid therapy dosage, timing and period after withdrawal in modulating the HSD11B2 promoter methylation status. It cannot be excluded, in fact, that different dosage or timing of administration or a free-from-steroid-therapy period might influence methylation at HSD11B2 promoter site. Since the significantly higher methylation was observed after one month from therapy, one could speculate that its drop after suspension may possibly occur even in a time shorter than one year.
The lack of statistical significance in the decrease of methylation values after prednisone withdrawal may also be related to the inherent stochastic nature of epigenetic processes [19] and, in the present study, to the relatively small number of participating subjects. Although the rather small number of patients studied is one of the limitations of this study, their homogeneity in terms of clinical characteristics, dosage and timing of prednisone utilized and time of analysis after withdrawal may give reason, at least partially, of our results in terms of functional epigenetic modulation. Indeed, a larger study is needed to confirm the present findings. Moreover, further ad hoc studies are certainly warranted to define the minimal dosage needed to modify the enzyme function by modulating HSD11B2 gene promoter methylation, and the required timing to reverse the phenomenon.
The methylation status observed at Region 1 of the HSD11B2 promoter confirms our previous results on the functional effect of methylation at this promoter region, results obtained with a different methodological approach [12].
As previously reported by others [16] we confirm the higher mean levels of methylation at HSD11B2 Region 2 as compared with Region 1 (Figure 2). The variations associated to prednisone therapy in Region 2 were similar, although not significant, to those observed in Region 1.
It could be speculated that the differential effect on methylation may be linked to other transcriptional regulatory factors at specific sites [20]. This hypothesis, however, needs further ad hoc studies.
Epigenetic mechanisms, including DNA methylation, are considered tissue-specific, however, several studies already showed that methylation status, either genomic or gene-specific, in PBMCs DNA may reflect a systemic epigenetic fingerprint [21]. These results are, therefore, of interest in the perspective of a potential use of PBMCs DNA methylation as a molecular biomarker for clinical purposes..
The mechanisms responsible of the higher HSD11B2 promoter methylation observed in prednisone-treated patients are yet matter of speculation. It is well known the link between DNA methylation and one-carbon metabolism for the provision of methyl groups by S-adenosylmethionine for the methylation reaction catalyzed by methyltransferases [22]. On another hand, glucocorticoids are able to up-regulate S-adenosylmethionine synthase activity [20,23,24]. In a rodent experimental model, a role for glucocorticoids in affecting one-carbon metabolism has been suggested [25-27] to modulate DNA methylation [28,29]. A number of mechanisms have been certainly claimed for being involved in the pathogenesis of glucocorticoid-induced hypertension [30] where a role for epigenetics can be also considered.
Epigenetic phenomena, among which DNA methylation, are lately emerging as potential important mechanisms for regulation of transcriptional expression of genes implied in complex, multifactorial diseases such as the case of arterial hypertension [3]. The observation of the functional role of methylation at HSD11B2 gene promoter and, furthermore, the induction and reversibility of this phenomenon in association with high-dose glucocorticoid therapy is a novel finding of potential high interest.
In conclusion, this study shows that 11beta-HSD2 is dynamically regulated during glucocorticoid therapy by methylation at HSD11B2 gene promoter site. The enzyme activity is functionally influenced as shown by the altered THFs/THE ratio, a surrogate biochemical marker of 11beta-HSD2 enzyme function. Remarkably, the epigenetic regulation of HSD11B2 is induced during prednisone therapy and reverses after prednisone withdrawal. The methylation status of HSD11B2 observed in PBMCs DNA may be considered as a useful epigenetic molecular marker of steroid-induced hypertension.
Conflict of Interest
The authors declare that they have no conflict of interest.
Acknowledgement
The work was performed in part in the LURM (Laboratorio Universitario di Ricerca Medica) Research Center, University of Verona.
References
- Ferrari P, Krozowski Z (2000) Role of the 11beta-hydroxysteroid dehydrogenase type 2 in blood pressure regulation. Kidney Int57:1374-1381.
- Carvajal CA, Romero DG, Mosso LM, Gonzalez AA, Campino C, et al (2005) Biochemical and genetic characterization of 11 beta-hydroxysteroid dehydrogenase type 2 in low-renin essential hypertensives. J Hypertens 23:71-77.
- Friso S, Carvajal CA, Fardella CE, Olivieri O (2015) Epigenetics and arterial hypertension: the challenge of emerging evidence. Transl Res 165:154-165.
- Naray-Fejes-Toth A, Watlington CO, Fejes-Toth G (1991) 11 beta-Hydroxysteroid dehydrogenase activity in the renal target cells of aldosterone. Endocrinology 129:17-21.
- Stewart PM, Corrie JE, Shackleton CH, Edwards CR (1988) Syndrome of apparent mineralocorticoid excess. A defect in the cortisol-cortisone shuttle. J Clin Invest 82:340-349.
- Kotelevtsev Y, Brown RW, Fleming S, Kenyon C, Edwards CR, et al. (1999) Hypertension in mice lacking 11beta-hydroxysteroid dehydrogenase type 2. J Clin Invest 103:683-689.
- Quinkler M, Zehnder D, Lepenies J, Petrelli MD, Moore JS, Hughes SV, et al. (2005) Expression of renal 11beta-hydroxysteroid dehydrogenase type 2 is decreased in patients with impaired renal function. Eur J Endocrinol 153:291-299.
- Wilson RC, Dave-Sharma S, Wei JQ, Obeyesekere VR, Li K, Ferrari P, et al. (1998) A genetic defect resulting in mild low-renin hypertension. Proc Natl Acad Sci U S A 95:10200-10205.
- Olivieri O, Pizzolo F, Ravagnani V, Moretti L, Carletto A, Faccini G et al. (2008) Urinary cortisol to cortisone metabolites ratio in prednisone-treated and spontaneously hypertensive patients. J Hypertens 26:486-493.
- Ferrari P, Sansonnens A, Dick B, Frey FJ (2001)In vivo 11beta-HSD-2 activity: variability, salt-sensitivity, and effect of licorice. Hypertension 38:1330-1336.
- Alikhani-Koopaei R, Fouladkou F, Frey FJ, Frey BM (2004) Epigenetic regulation of 11 beta-hydroxysteroid dehydrogenase type 2 expression. J Clin Invest 114:1146-1157.
- Friso S, Pizzolo F, Choi SW, Guarini P, Castagna A, Ravagnani V, et al. (2008) Epigenetic control of 11 beta-hydroxysteroid dehydrogenase 2 gene promoter is related to human hypertension. Atherosclerosis 199:323-327.
- Olivieri O, Ciacciarelli A, Signorelli D, Pizzolo F, Guarini P, Pavan C, et al. (2004) Aldosterone to Renin ratio in a primary care setting: the Bussolengo study. J Clin Endocrinol Metab 89:4221-4226.
- Shackleton CH (1993) Mass spectrometry in the diagnosis of steroid-related disorders and in hypertension research. J Steroid Biochem Mol Biol 45:127-140.
- Campino C, Carvajal CA, Cornejo J, San Martin B, Olivieri O, Guidi G, et al. (2010) 11beta-Hydroxysteroid dehydrogenase type-2 and type-1 (11beta-HSD2 and 11beta-HSD1) and 5beta-reductase activities in the pathogenia of essential hypertension. Endocrine 37:106-114.
- Zhao Y, Gong X, Chen L, Li L, Liang Y, Chen S, et al. (2014) Site-specific methylation of placental HSD11B2 gene promoter is related to intrauterine growth restriction. Eur J Hum Genet 22:734-740.
- Geraci M (2014) Linear Quantile Mixed Models: The lqmm Package for Laplace Quantile Regression. Journal of Statistical Software 57:1-29.
- Jones PA (1999) The DNA methylation paradox. Trends Genet 15:34-37.
- Bjornsson HT, Fallin MD, Feinberg AP (2004) An integrated epigenetic and genetic approach to common human disease. Trends Genet 20:350-358.
- Bing Y, Zhu S, Yu G, Li T, Liu W, et al. (2014) Glucocorticoid-induced S-adenosylmethionine enhances the interferon signaling pathway by restoring STAT1 protein methylation in hepatitis B virus-infected cells. J Biol Chem 289:32639-32655.
- Friso S, Udali S, Guarini P, Pellegrini C, Pattini P, Moruzzi S, et al. Global DNA hypomethylation in peripheral blood mononuclear cells as a biomarker of cancer risk. Cancer Epidemiol Biomarkers Prev 2013.
- Friso S, Choi SW (2005) Gene-nutrient interactions in one-carbon metabolism. Curr Drug Metab 6:37-46.
- Gil B, Pajares MA, Mato JM, Alvarez L (1997) Glucocorticoid regulation of hepatic S-adenosylmethionine synthetase gene expression. Endocrinology 138:1251-1258.
- Garcia-Trevijano ER, Latasa MU, Carretero MV, Berasain C, Mato JM, et al. (2000) S-adenosylmethionine regulates MAT1A and MAT2A gene expression in cultured rat hepatocytes: a new role for S-adenosylmethionine in the maintenance of the differentiated status of the liver. Faseb J 14:2511-2518.
- Kim MH, Kim E, Passen EL, Meyer J, Kang SS (1997) Cortisol and estradiol: nongenetic factors for hyperhomocyst(e)inemia. Metabolism 46:247-249.
- Rowling MJ, Schalinske KL (2003) Retinoic acid and glucocorticoid treatment induce hepatic glycine N-methyltransferase and lower plasma homocysteine concentrations in rats and rat hepatoma cells. J Nutr 133:3392-3398.
- Enokido Y, Suzuki E, Iwasawa K, Namekata K, Okazawa H, Kimura H (2005) Cystathionine beta-synthase, a key enzyme for homocysteine metabolism, is preferentially expressed in the radial glia/astrocyte lineage of developing mouse CNS. FASEB J 19:1854-1856.
- Friso S, Choi SW, Girelli D, Mason JB, Dolnikowski GG, Bagley PJ, et al. (2002) A common mutation in the 5,10-methylenetetrahydrofolate reductase gene affects genomic DNA methylation through an interaction with folate status. Proc Natl Acad Sci USA 99:5606-5611.
- Friso S, Choi SW (2002) Gene-nutrient interactions and DNA methylation. J Nutr 132:2382S-2387S.
- Brem AS (2001) Insights Into Glucocorticoid-Associated Hypertension. Am J Kidney Dis 37:1-10.

