Keywords
Cystic fibrosis; Pancreatitis; CFTR; Therapy; Pancreatic
insufficiency
METHODS
We performed a comprehensive literature search using
the electronic Pubmed database for relevant studies on
cystic fibrosis and pancreatic-related disorders. Relevant
MeSH keywords were used (“cystic fibrosis, pancreatitis,
CFTR, therapy, pancreatic insufficiency”). We limited the
search to English-language articles published from January
1, 2010 to April 1, 2021. Two independent reviewers
assessed the articles for eligibility. Additional relevant
studies that were identified upon manual search of the
bibliographies of the primary articles, though outside the
time window, were later included in this review. Studies
published before January 2000 were excluded from the
review.
RESULTS
Seventy-eight abstracts overall were retrieved from
our literature search. After appraisal by two investigators,
including addition of relevant studies from bibliographies
of the primary articles, fifty-six publications were included
in our synthesis and narrative literature review.
DISCUSSION
Pathophysiology
Pancreatic ductal secretion plays a vital role in healthy
exocrine function of the pancreas [1]. The ductal cells
produce a bicarbonate-rich secretion that promotes
an ideal alkaline environment for zymogens, prevents
premature inactivation of digestive enzymes, and washes
out potential factors like bile acid from the biliary tree
[2]. Specifically, the CFTR protein regulates chloride and
bicarbonate transport across epithelial cell membranes
into the lumen, as well as the pH of the epithelial secretions
[3]. The WNK1 (with-no-lysine kinase 1) regulates
permeability of the CFTR via an intracellular chlorideconcentration
dependent association [4]. The CFTR
channel is either absent or dysfunctional in secretory
gland diseases like cystic fibrosis [5], which causes
decreased ductal bicarbonate secretion and the formation
of viscous plugging, as well as a low pH environment which
can precipitate premature enzyme activation, scarring,
and eventual pancreatic insufficiency [5]. Moreover,
premature activation of trypsinogen into trypsin plays
an important role in the decreased ductal secretion via
inhibition of Cl-/HCO3- exchangers and CFTR Cl- channels,
thereby perpetuating the cycle of ductal obstruction and
inflammation.
Abnormal or absent CFTR also has implications for
acinar cells. Ductal inflammation, as a consequence of
viscous plugging from deficient ductal secretion, has
shown to inhibit acinar cell secretion, promote acinar cell degradation, and ultimately lead to parenchymal damage
[6]. It is the initial insult to the ducts, then, that may be
causing secondary downstream damage to the acinar
cells [6]. In addition to the HCO3- mechanism, aquaporins
have been investigated for their role in pancreatic fluid
formation and pancreatitis. Aquaporins are membrane
proteins that mediate transcellular water transport. In
particular, aquaporin 1 co-localizes with the CFTR channel
in the apical membrane of the pancreatic ductal cells [2].
Damaged CFTR reduces the water permeability of ductal
cells via decreased expression of aquaporin, causing
reduced ductal fluid and HCO3-secretion overall [2]. Thus,
CFTR plays an important role in the pathophysiology of
aquaporins and development of pancreatic inflammation,
evidenced by the decreased expression of aquaporins in
acute and chronic pancreatitis.
CFTR is also responsible for intracellular signaling in
pancreatic epithelial cells via calcium. The loss of CFTR
has therefore been implicated in excess mitochondrial
calcium accumulation, increased permeability of the inner
mitochondrial membrane, and subsequent mitochondrial
swelling, apoptosis and necrosis [7].
In addition to protein channels, pancreatitis-inducing
factors and toxins also modulate CFTR expression
and activity (Table 1). Ethanol and fatty acids have
demonstrated a dose-dependent inhibition of CFTR
and decreased fluid and HCO3- secretion as a result [3].
Moreover, cigarette smoke increases intracellular calcium
and causes downstream CFTR channel internalization and
down-regulation of CFTR expression, as well as increased
sweat chloride levels [8, 9]. Some studies speculate that constituents of smoke like acrolein, cadmium, and
manganese -and specifically the free radical-induced
damage by smoke-may also directly be responsible for
inhibition of CFTR [10].
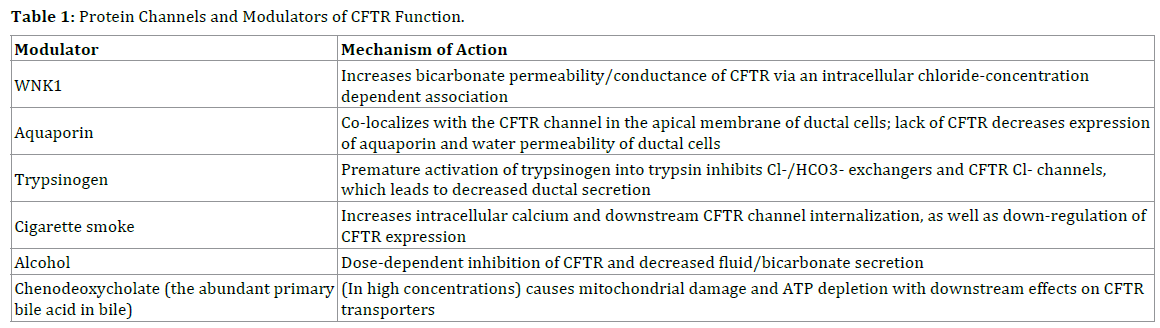
In regard to biliary pancreatitis, chenodeoxycholate, the
abundant primary bile acid in bile, in high concentrations
has shown to cause mitochondrial damage and ATP
depletion with downstream effects on CFTR transporters
[11, 12]. Thus pancreatic ductal secretion of bicarbonate is
strongly inhibited [3].
Pancreatic insufficiency
Historically, pancreatic insufficiency was associated
with significant early childhood mortality before the
advent of pancreatic enzyme replacement therapy and
improved nutritional care. The vast majority (80%)
of cystic fibrosis patients are pancreatic-insufficient
[13], largely due to ductal plugging and autodigestion
of the pancreas throughout the embryonic stage [14].
Once patients have sufficient pancreatic damage to the
extent of losing exocrine function, the pancreas becomes
functionally inept and lack the acinar reserve to develop
pancreatitis (Figure 1).
Figure 1: Axial CT image of a 19 year-old patient with one copy of a CFTR mutation with near complete fatty replacement of the pancreas and symptoms of pancreatic exocrine insufficiency.
Pancreatic exocrine sufficiency is affected by the
presence of either severe or mild mutation on each
respective CFTR allele [3]. Patients with the mild CFTR
genotype are often pancreatic sufficient but have increased
risk for pancreatitis [15]. Pancreatic-sufficient patients also
are at increased risk for developing exocrine insufficiency,
once they have had an index episode of pancreatitis
[15]. Patients with pancreatic insufficiency typically develop malabsorption symptoms of foul-smelling stools,
abdominal discomfort and weight loss. Combined with the
highly catabolic process from pancreatic inflammation and
autodigestion, pancreatic insufficiency when left untreated
can progress to severe malnutrition and growth failure.
Malnutrition in pancreatic insufficiency is multifactorial:
malabsorption from deficient pancreatic enzymes, bile
salt precipitation impairing lipid solubilization, altered
gut motility and mucosal uptake, inadequate energy
intake, and increased energy requirements [14]. In the
appropriate clinical context, indirect pancreatic function
tests like a pancreatic fecal elastase help make a diagnosis
of pancreatic insufficiency with symptoms and signs
suggestive of malabsorption. A 72 hour fecal fat test
remains the gold standard test to diagnose pancreatic
insufficiency, though this can be cumbersome to carry out
in clinical practice.
Pancreatic enzyme replacement therapy in cystic
fibrosis
Standard of care therapy for pancreatic insufficiency
is pancreatic enzyme replacement therapy (PERT) with
fat-soluble vitamin supplementation. PERT contains the
exogenous lipase, amylase, and protease that digests their
respective macromolecules, which is vital in patients
with cystic fibrosis for normal growth and nutrition.
Without PERT, patients are at risk for severe lung disease,
distal intestinal obstruction syndrome, and decreased
life expectancy. PERT formulations are porcine-derived
and either enteric-coated (which are protected against
denaturing by gastric acid) or non-enteric coated.
Microsphere formulations, which allow for smaller size
preparation and more stable delivery, theoretically hasten
delivery of the enzymes into the duodenum, but clinical
trials have failed to demonstrate substantial improvement
in bowel symptoms with microsphere formulations
[16]. Moreover, the type of preparation has not shown
significant differences on bowel symptoms, quality of life,
or lung disease outcomes [16].
Pancreatic insufficiency is frequently underdiagnosed
and PERT underdosed, and yet there remains significant
heterogeneity between cystic fibrosis centers in regard to
management of PERT. Some neonatal screening programs
will administer PERT within the first few weeks of a
diagnosis of cystic fibrosis; some centers will initiate PERT
in clinical scenarios of growth failure in children and adults
in failure to thrive, whereas others will rely on pancreatic
function testing to guide decision-making on starting PERT
[16]. Myriad factors contribute to the efficacy of PERT in
each patient: the residual pancreatic function, meal size and
fat content, and level of bile acid secretion and intestinal pH
[17]. Titration of PERT is largely tailored to each individual
patient. Improvement in steatorrhea should be assessed,
as should surrogate markers such as fat-soluble vitamin
levels, nutrition indices, and quality of life metrics [18];
fecal elastase is unaffected by PERT and should not be used
to assess clinical response. Side effects of PERT can include
nausea, constipation, diarrhea, abdominal cramping, bloating and less commonly, fibrosing colonopathy with
high doses of PERT exceeding 10000 units per kilogram
body weight per day [16, 17].
Acute pancreatitis
The prevalence of acute pancreatitis in patients with
cystic fibrosis overall is estimated to be 0.5-1.7%, though
patients with pancreatic sufficiency have a higher risk
(32%) of developing pancreatitis [19]. Mutation of the
CFTR genotype is one predisposing factor believed to be
responsible for this increased risk. CFTR mutations have
been speculated to induce acute pancreatitis in two ways:
1) decrease in pancreatic fluid secretion thereby producing
more viscosity and distal obstruction of the duct, and 2)
development of excess inflammatory response [20]. The
type of CFTR mutation significantly correlates with the
risk of acute pancreatitis. And it is the mild CFTR genotype
that has a dominant phenotypic affect, compared to that of
the severe genotype [15].
It remains unclear why there is such heterogeneity in
which pancreatic-sufficient patients develop pancreatitis.
Why patients with identical CFTR genotypes can have such
variable presentations of pancreatitis is also unclear [19].
One hypothesis is that other non-CFTR genetic variants
or environmental exposures (ie, tobacco, alcohol) may be
additional predisposing factors for the development of
pancreatitis. However, a prospective study of pancreaticsufficient
patients [19] showed that while the majority of
mild mutations were found in the first transmembrane
domain, there was no direct correlation between non-
CFTR genotypes (including SPINK1, PRSS1, CTRC, CASR,
and SLC26A6) and the prevalence of acute or recurrent
pancreatitis. Yet it remains very plausible that multiple
players including these genetic variants and environmental
exposures work synergistically to contribute to
pancreatitis, and further mechanistic studies are needed
to better understand and model the interactions between
these cofactors [3, 19, 21].
Pancreas Divisum
Pancreas divisum is a variant anatomy of the pancreatic
duct resulting from failed fusion of the ventral and dorsal
pancreatic buds during embryonic development, leading
to pancreatic drainage thru the minor papilla (Figure 2).
There is some controversy whether pancreas divisum
is a true risk factor for pancreatitis or a nonpathological
clinical entity. Although pancreas divisum is surprisingly
prevalent (up to 5-10% in autopsy series), only a minority
of patients with divisum develop pancreatitis [20]. Studies
have failed to consistently show increased frequency of
pancreatitis in patients with pancreas divisum, when
compared to controls [22]. These findings suggest that
pancreas divisum per se may not cause pancreatitis.
However, pancreas divisum has shown to be more
frequent in cases of genetic pancreatitis, particularly
CFTR mutations [22]. CFTR mutations have independently
been associated with pancreatic ductal obstruction and
inflammation. As pancreas divisum theoretically can lead to increased intraductal pressure by way of drainage
through the smaller minor papilla, pancreas divisum may
ultimately serve as a cofactor with CFTR in the development
of pancreatitis [23]. On the other hand, most cystic fibrosis
patients are pancreatic insufficient and are less likely
to have develop pancreatitis. It is postulated, then, that
pancreas divisum may be protective against pancreatic
insufficiency in CFTR carriers, at the cost of increased risk
for pancreatitis [24]. Further studies are needed to assess
whether or not surgical or endoscopic interventions to
enlarge the minor papilla orifice reduces the likelihood of
pancreatitis in patients with pancreas divisum and CFTR
mutation.
Figure 2: Complete pancreas divisum seen on an MRCP in patient with one copy of a CTFR mutation and recurrent acute pancreatitis.
Chronic pancreatitis
Chronic pancreatitis is characterized by progressive
inflammation of the pancreas and ductal plugging [25],
which leads to acinar loss, lobular and periductal fibrosis,
and irreversible pancreatic damage [26]. These changes
are due to dysregulation of HCO3- secretion. However,
morphometric analysis of pancreatic ductal mucosa has
also shown increased mucus content in the small pancreatic
ducts in chronic pancreatitis, as well as increased expression
of secreted mucins MUC5B and MUC6 [25]. Whether
the mucus hypersecretion via differential expression of
mucin proteins is a result or cause of the increased mucus
content and associated plugging in the pancreatic ducts
remains unclear [25]. Therapies that increase ductal
hydration and improve mucus clearance, therefore, may
play a vital role in washing out the digestive enzymes and
toxins in the early stages of chronic pancreatitis, before
progressive and irreversible inflammation develop [27].
Genetic variants including PRSS1, SPINK1, CTRC, and
CFTR have long been associated with the development
of chronic pancreatitis, presumably to their effect of
premature trypsinogen activation [3, 28, 29]. Although
CPA1 has previously been implicated in the pathogenesis
of chronic pancreatitis [30], Hegyi et al. [31] was the first
in-vivo study to demonstrate CPA1 as a disease-modifying
factor for chronic pancreatitis, via enzyme misfolding and
induction of endoplasmic reticulum stress. This pathway
may ultimately have therapeutic implications which are independent of the trypsinogen pathway. There are also
functional CFTR variants with impaired HCO3- (but not
Cl-) permeability which can predispose patients to chronic
pancreatitis, but not cystic fibrosis [5, 32].
Pancreatic cystosis
Pancreatic cystosis in cystic fibrosis is relatively
uncommon, and large cysts spanning greater than 1
centimeter are even more rare, occurring in only 8% of
patients with cystic fibrosis [33]. Little is known regarding
the natural history and clinical outcomes of pancreatic
cystosis. A recent systematic review showed that
symptoms on initial presentation were noted in only 42%
of total reported cases, with no strong correlation between
size of cyst and symptoms [34]. There is neither proven
risk of progression to malignancy with pancreatic cystosis
[35] nor copious information on the natural history
of cytosis, so it is reasonable to hold off on treatment
if patients are asymptomatic. Decisions on therapy,
therefore, are predicated on factors such as severity of pain
symptoms, quality of life, obstructive symptoms, as well as
comorbidities such as recurrent pulmonary complications
[34].
CF related diabetes
Cystic fibrosis-related diabetes is a pancreatic
complication that increases in prevalence with age,
up to 40-50% of patients older than 40 years of age
[36]. Progressive damage to the pancreatic islet cells
leads to insulin deficiency, contributing to diabetes.
Other factors that can impair glucose tolerance include
infection, malnutrition, concomitant liver disease, and
bacterial overgrowth [14]. Diabetes in turn can also
have a multifactorial effect on pancreatic duct secretion:
decreased or absent endogenous insulin impairing
secretion at both the ductal (decreased duct morphology)
and acinar level (tissue atrophy), autoantibodies against
carbonic anhydrase, autonomic neuropathic effect on
ductal secretion, negative feedback of elevated blood
glucose on sodium-dependent glucose transporter and
downstream bicarbonate secretion, and decreased Na+/
K+-ATPase activity [5]. Cystic fibrosis-related diabetes is associated with increased mortality, decreased pulmonary
function and malnutrition [36]. Moreover, screening and
detection can be challenging particularly in the early
stages of cystic fibrosis-related diabetes, as these patients
may be at risk for microvascular complications well before
traditional tests like HbA1c detect diabetic disease [36].
Typically, once a diagnosis of cystic-fibrosis related disease
is confirmed via oral glucose tolerance test, basal insulin
and adequate caloric/energy intake is recommended with
the supervision of a multidisciplinary team with a cystic
fibrosis dietician.
Advances in CFTR modulators and other therapies
Initial therapies for cystic fibrosis were largely
supportive including inhaled mucolytics, antibiotics, and
pancreatic enzyme replacement therapy. Ideally to halt
disease progression, treatments that target the underlying
CF mutation present the most promise. There are over
two thousand known CFTR mutations, though most have
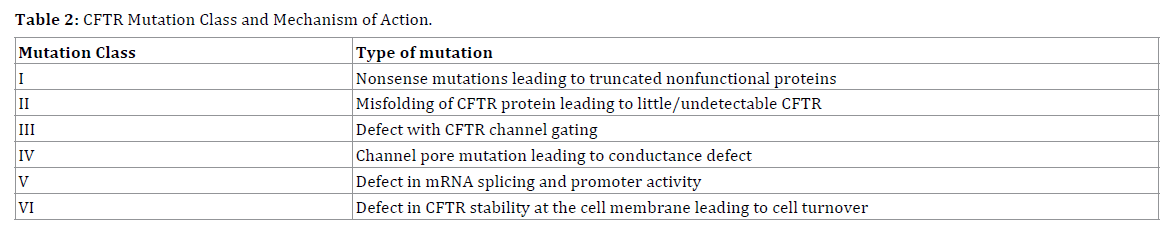
no clinically significant phenotype. The mutations that are
most typically associated with disease are categorized class
I-VI mutations (Table 2). Class I mutation results in truncated
nonfunctional proteins and Class II non-expressed CFTR
protein at the plasma membrane [37]. F508del is in class
II and the most common CFTR mutation (up to 80-90% in
patients with cystic fibrosis) [38]. For classes III and IV, the
CFTR protein is fully synthesized and expressed, but there are
defects with CFTR channel gating (III) and conductance via
channel pore defect (IV). Class V mutation is a defect in mRNA
splicing and promoter activity, whereas Class VI mutation
deals with defective CFTR stability at the cell membrane and
resultant cell turnover [37].
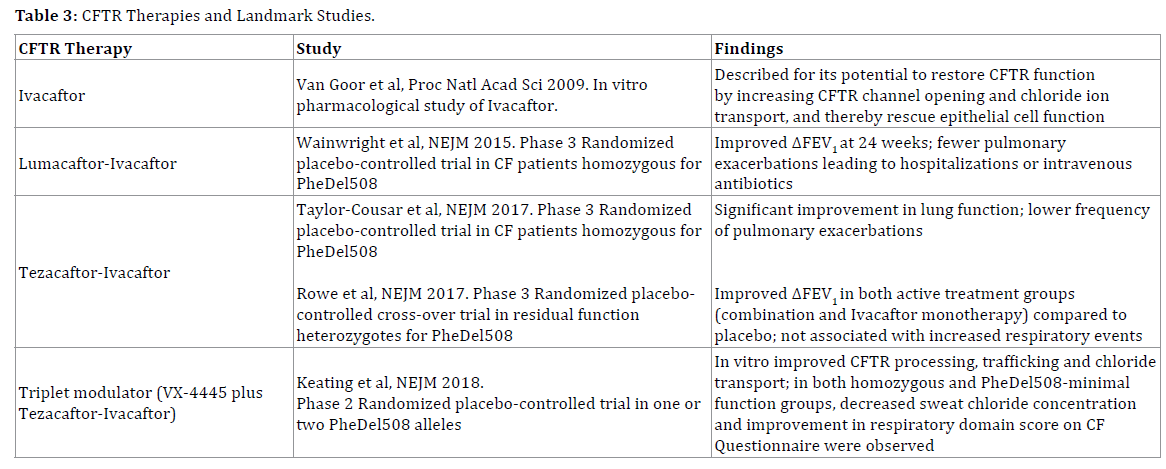
Therapeutic efforts in cystic fibrosis therefore have
been directed at these specific mutations (Table 3).
Ivacaftor was first identified as a CFTR potentiator that
increases the CFTR channel opening at the cell surface and
thereby enhances chloride ion transport [39]. It went on
to demonstrate normalized fecal elastase and decreased
number of pancreatitis episodes in patients with CFTR
dysfunction [40]. Moreover, ivacaftor has demonstrated
in vivo evidence of improved proximal small intestinal
pH, which supports the hypothesis of CFTR modulator’s
effect on bicarbonate secretion, gut hydration, and gut
biosis as well as downstream effects on weight gain [41].
However, ivacaftor has not shown to be effective in a
significant portion of the cystic fibrosis population, namely
the F508del homozygous patients [42]. CFTR correctors
were also explored, which increase trafficking of the
CFTR protein to the cell surface. The corrector lumacaftor,
though shown to improve CFTR function in the sweat
duct, failed as monotherapy in demonstrating significant
improvement in outcomes like lung function [43].
The question then arose whether combining a CFTR
corrector with a potentiator may provide added benefit.
CFTR correctors and potentiators have been investigated
in combination in mouse models for their role in rescuing
ductal CFTR. In mouse models of Sjogren’s and chronic
pancreatitis, CTFR potentiator VX770 and corrector C18
restored ductal CFTR, as well as increased calcium signaling,
aquaporin expression, and fluid secretion in acinar cells [6].
Lumacaftor-ivacaftor was first approved for combination
use after demonstrating modest improvement, compared
to placebo, in FEV1 (forced expiratory volume during
first second) of patients with cystic fibrosis homozygous for PheDel508 CFTR [44]. Because of the modest effect,
potential drug interaction between lumacaftor and
ivacaftor, and side effects of dyspnea and liver damage,
another combination tezacaftor-ivacaftor was investigated
and found to have similar effect size with lung function,
albeit improved side effect profile [45, 46].
Further studies have since investigated triplet CFTR
modulator therapy: two correctors with distinct binding
sites and mechanisms are hypothesized to increase
PheDel508 processing and trafficking to the cell surface,
which can then be potentiated by a third agent like ivacaftor
[47]. Tezacaftor-ivacaftor has served as the foundation for
combination therapy with next-generation correctors such
as VX-440, VX-152, and VX-659 [48], with randomized trial
studies demonstrating significant improvement in FEV1
[47, 49].
Although triplet modulator therapy with novel
correctors show promise for the PheDel508 patients
which make up the bulk of the CF population, up to 8-10%
remaining have nonsense mutations resulting in premature
termination of CFTR protein synthesis [50]. Additional
therapies requiring ongoing study include eluforsen, an
antisense oligonucleotide that repairs the genetic defect
in RNA, which has demonstrated safety and efficacy with
respiratory symptoms in phase 1 trials [51]. Ataluren had
been studied for its potential benefit in nonsense mutation
cystic fibrosis via reading through premature termination
codons and producing functional CFTR proteins—but
results in phase 3 have failed to show significant pulmonary
benefit from placebo [52]. Gene editing strategies have
been explored with recent studies using Cas9 and adenoassociated
virus 6 to correct CFTR mutation in upperairway
basal cell stem cells ex vivo [53]. These corrected
basal cells can then be embedded onto a scaffold for
engraftment while retaining their differentiation capacity.
Other approaches being investigated include derivation
of donor-specific induced pluripotent cells with CFTR
correction and directed differentiation to airway basal
cells, as well as direct in vivo editing of airway cells [54].
Such gene editing and replacement therapy may ultimately
hold promise for the patients who do not respond to
triplet modulator therapy. An additional challenge will
be the selection of more sensitive clinical endpoints, as
traditional outcomes of FEV1 and number of pulmonary
exacerbations [1] have significantly improved with
triplet modulator therapy. Future clinical trials will need
endpoints that better encompass pancreatic function and
end-stage complications.
TPIAT in CF patients
TPIAT (total pancreatectomy with islet
autotransplantation) is an appropriate treatment option
for severe chronic pancreatitis and recurrent acute
pancreatitis. It can relieve pain as well as preserve betacell
mass to reduce the risk of post-operative diabetes
[55]. Moreover, when compared to controls without
CFTR mutations, patients with CFTR homozygotes and
heterozygote mutations who underwent TPIAT had similar post-operative Hgb A1c, C-peptide, islet yield, and
post-operative complications [55]. TPIAT may be a viable
treatment option in cystic fibrosis patients with medically
refractory chronic pancreatitis and recurrent acute
pancreatitis. Also TPIAT has demonstrated substantive
pain relief and improved quality of life for pancreatic
cystosis in cystic fibrosis [56]. Provided that islet cells are
preserved, and cysts drained prior to digestion to ensure
a high islet yield, TPIAT can provide a surgical option for
these difficult cases. Overall further studies are needed to
risk stratify patients undergoing TPIAT with concomitant
lung and pancreas involvement of cystic fibrosis.
CONCLUSIONS
CFTR mutation in cystic fibrosis leads to ductal plugging,
pancreatic inflammation and downstream pancreatic
insufficiency which can cause severe malnutrition and
growth failure. PERT is vital in the management of
pancreatic insufficiency, nutrition and overall morbidity in
patients with cystic fibrosis. Trypsinogen and aquaporins
may play an important role in the pathophysiology of
pancreatic ductal secretion. The development of acute
pancreatitis depends on cofactors of genetic variants,
environmental exposures, and severity of CFTR genotype.
Other pancreatic complications in cystic fibrosis include
cystic fibrosis-related diabetes and, less commonly,
pancreatic cystosis; pancreatic divisum may be a cofactor
for pancreatitis rather than an independent risk factor per
se. Therapies for cystic fibrosis have evolved with triplet
modulator therapy demonstrating the potential to alter
disease trajectory in the majority of cystic fibrosis patients.
However, ongoing investigation with gene replacement
therapy and antisense oligonucleotide therapy will be
necessary to meet the needs of patients with nonsense
mutations, who may not respond to triplet modulator
therapy.
ACKNOWLEDGMENT
The authors received no financial support for the
research, authorship, and/or publication of this article. The
authors have no competing financial interests or personal
relationships to disclose.
Conflicts of Interest
The authors declare no competing interest.
References
- Elborn JS. Cystic fibrosis. Lancet 2016; 388:2519-2531. [PMID: 27140670].
- Venglovecz V, Pallagi P, Kemény LV, Balázs A, Balla Z, Becskeházi E, et al. The importance of aquaporin 1 in pancreatitis and its relation to the CFTR Cl(-) Channel. Front Physiol 2018; 9:854. [PMID: 30050452].
- Hegyi P, Wilschanski M, Muallem S, Lukacs GL, Tóth MS, Aliye UC, et al. CFTR: A new horizon in the pathomechanism and treatment of pancreatitis. Rev Physiol Biochem Pharmacol 2016; 170:37-66. [PMID: 26856995].
- Yonjung Kim, Ikhyun Jun, Shin DH, Jihoon G Yoon, He Piao, Jinsei Jung, et al. Regulation of CFTR bicarbonate channel activity by wnk1: implications for pancreatitis and CFTR-related disorders. Cell Mol Gastroenterol Hepatol 2020; 9:79-103. [PMID: 31561038].
- Pallagi P, Hegyi P, Rakonczay Z. The physiology and pathophysiology of pancreatic ductal secretion: The background for clinicians. Pancreas 2015; 44:1211-33. [PMID: 26465950].
- Zeng M, Szymczak M, Ahuja M, Zheng C, Yin H, Swaim W, et al. Restoration of CFTR activity in ducts rescues acinar cell function and reduces inflammation in pancreatic and salivary glands of mice. Gastroenterology 2017; 153:1148-1159. [PMID: 28634110].
- Madácsy T, Pallagi P, Maleth J. Cystic fibrosis of the pancreas: The role of cftr channel in the regulation of intracellular Ca(2+) signaling and mitochondrial function in the exocrine pancreas. Front Physiol. 2018; 9:1585. [PMID: 30618777].
- Rasmussen JE, Sheridan JT, Polk W, Davies CM, Tarran R. Cigarette smoke-induced Ca2+ release leads to cystic fibrosis transmembrane conductance regulator (CFTR) dysfunction. J Biol Chem 2014; 289:7671-81. [PMID: 24448802].
- Raju SV, Jackson PL, Courville CA, McNicholas CM, Sloane PA, Sabbatini G, et al. Cigarette smoke induces systemic defects in cystic fibrosis transmembrane conductance regulator function. Am J Respir Crit Care Med 2013; 188:1321-30. [PMID: 24040746].
- Hassan F, Xu X, Nuovo G, Killilea DW, Tyrrell J, Tan CD, et al. Accumulation of metals in GOLD4 COPD lungs is associated with decreased CFTR levels. Respir Res 2014; 15:69. [PMID: 24957904].
- Venglovecz V, Rakonczay Z, Ozsvári B, Takács T, Lonovics J, Varró A, et al. Effects of bile acids on pancreatic ductal bicarbonate secretion in guinea pig. Gut 2008; 57:1102-12. [PMID: 18303091].
- Maléth J, Venglovecz V, Rázga Z, Tiszlavicz L, Rakonczay Z, Hegyi P. Non-conjugated chenodeoxycholate induces severe mitochondrial damage and inhibits bicarbonate transport in pancreatic duct cells. Gut 2011; 60:136-8. [PMID: 20732916].
- Carol Durno, Mary Corey, Julian Zielenski, Elizabeth Tullis, Lap-Chee Tsui, Peter Durie. Genotype and phenotype correlations in patients with cystic fibrosis and pancreatitis. Gastroenterology 2002; 123:1857-64. [PMID: 12454843].
- M Sinaasappel, M Stern, J Littlewood, S Wolfe, G Steinkamp, Harry G M Heijerman, et al. Nutrition in patients with cystic fibrosis: a European Consensus. J Cyst Fibros 2002; 1:51-75. [PMID: 15463811].
- Ooi CY, Dorfman R, Cipolli M, Gonska T, Castellani C, Keenan K, et al. Type of CFTR mutation determines risk of pancreatitis in patients with cystic fibrosis. Gastroenterology 2011; 140:153-61. [PMID: 20923678].
- Somaraju URR, Moya AS. Pancreatic enzyme replacement therapy for people with cystic fibrosis. Cochrane Database Syst Rev 2020; 8:CD008227. [PMID: 32761612].
- Brennan GT, Saif MW. Pancreatic Enzyme Replacement Therapy: A Concise Review. JOP 2019; 20:121-125. [PMID: 31736680]
- Perbtani Y, Forsmark CE. Update on the diagnosis and management of exocrine pancreatic insufficiency. F1000Res 2019; 8:F1000 Faculty Rev-1991. [PMID: 31824646].
- Gaitch N, Hubert D, Gameiro C, Burgel PR, Houriez F, Martinez B, et al. CFTR and/or pancreatitis susceptibility genes mutations as risk factors of pancreatitis in cystic fibrosis patients? Pancreatology 2016; 16:515-22. [PMID: 27086061].
- Gelrud A, Sheth S, Banerjee S, Weed D, Shea J, Chuttani R, et al. Analysis of cystic fibrosis gener product (CFTR) function in patients with pancreas divisum and recurrent acute pancreatitis. Am J Gastroenterol 2004; 99:1557-62. [PMID: 15307877].
- Anita Balázs, Péter Hegyi. Cystic fibrosis-style changes in the early phase of pancreatitis. Clin Res Hepatol Gastroenterol 2015; 1:S12-7. [PMID: 26206571].
- Bertin C, Pelletier CL, Vullierme MR, Bienvenu T, Rebours V, Hentic O, et al. Pancreas divisum is not a cause of pancreatitis by itself but acts as a partner of genetic mutations. Am J Gastroenterol 2012; 107:311-7. [PMID: 22306946].
- Dray X, Fajac I, Bienvenu T, Chryssostalis A, Sogni P, Hubert D. Association of pancreas divisum and recurrent acute pancreatitis with the IVS8-5T-12TG allele of the CFTR gene and CFTR dysfunction. Pancreas 2007; 35:90-3. [PMID: 17575549].
- Nicholson JA, Johnstone M, Greenhalf W. Divisum may be preserving pancreatic function in CFTR patients-but at a cost. Am J Gastroenterol 2012; 107:1758-9. [PMID: 23160301].
- Balázs A, Balla Z, Kui B, Maléth J, Rakonczay Z, Duerr J, et al. Ductal Mucus Obstruction and Reduced Fluid Secretion Are Early Defects in Chronic Pancreatitis. Front Physiol 2018; 9:632. [PMID: 29896115].
- Joan M Braganza, Stephen H Lee, Rory F McCloy, Michael J McMahon. Chronic pancreatitis. Lancet 2011; 377:1184-97. [PMID: 21397320].
- Hegyi P, Petersen OH. The exocrine pancreas: the acinar-ductal tango in physiology and pathophysiology. Rev Physiol Biochem Pharmacol. 2013; 165:1-30. [PMID: 23881310].
- Witt H, Luck W, Hennies HC, Classen M, Kage A, Lass U, Landt O, Becker M. Mutations in the gene encoding the serine protease inhibitor, Kazal type 1 are associated with chronic pancreatitis. Nat Genet. 2000; 25:213-6. [PMID: 10835640].
- Rosendahl J, Witt H, Szmola R, Bhatia E, Ozsvári B, Landt O, et al. Chymotrypsin C (CTRC) variants that diminish activity or secretion are associated with chronic pancreatitis. Nat Genet. 2008; 40:78-82. [PMID: 18059268].
- Heiko Witt, Sebastian Beer, Jonas Rosendahl, Jian-Min Chen, Giriraj Ratan Chandak, Atsushi Masamune, et al. Variants in CPA1 are strongly associated with early onset chronic pancreatitis. Nat Genet 2013; 45:1216-20. [PMID: 23955596].
- Hegyi E, Sahin-Tóth M. Human CPA1 mutation causes digestive enzyme misfolding and chronic pancreatitis in mice. Gut 2019; 68:301-312. [PMID: 30045879].
- LaRusch J, Jung J, General IJ, Lewis MD, Park HW, Brand RE, et al. Mechanisms of CFTR functional variants that impair regulated bicarbonate permeation and increase risk for pancreatitis but not for cystic fibrosis. PLoS Genet 2014; 10:e1004376. [PMID: 25033378].
- de Vries JM, Green D, Kucera JN, Fabbrini AL, Kidder M, Brown J, Wilsey M. Cystic fibrosis-related pancreatic cysts decrease in size and number upon treatment with cystic fibrosis transmembrane conductance regulator modulators. Pancreas 2020; 49:e50-e51. [PMID: 32590622].
- Desai CS, Vonderau JS, McCall R, Khan KM, Baron TH. Pancreatic cystosis in patients with cystic fibrosis: A qualitative systematic review. Pancreatology 2018; 18:700-704. [PMID: 30139657].
- Burt H, Andronikou S, Langton-Hewer S. Pancreatic cystosis in cystic fibrosis. BMJ Case Rep. 2016; 2016:bcr2015214288. [PMID: 26944373].
- Plant BJ, Goss CH, Plant WD, Bell SC. Management of comorbidities in older patients with cystic fibrosis. Lancet Respir Med 2013; 1:164-74. [PMID: 24429097].
- Ponzano S, Nigrelli G, Fregonese L, Eichler I, Bertozzi F, Bandiera T, et al. A European regulatory perspective on cystic fibrosis: current treatments, trends in drug development and translational challenges for CFTR modulators. Eur Respir Rev 2018; 27:170124. [PMID: 29653946].
- Riordan JR. CFTR function and prospects for therapy. Annu Rev Biochem 2008; 77:701-26. [PMID: 18304008].
- Goor FV, Hadida S, Grootenhuis PD, Burton B, Cao D, Neuberger T, et al. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc Natl Acad Sci U S A. 2009; 106:18825–18830. [PMID: 19846789].
- Ilias Kounis, Philippe Lévy, Vinciane Rebours. Ivacaftor CFTR potentiator therapy is efficient for pancreatic manifestations in cystic fibrosis. Am J Gastroenterol 2018; 113:1058-1059. [PMID: 29887601].
- Daniel Gelfond, Sonya Heltshe, Changxing Ma, Steven M Rowe, Carla Frederick, Ahmet Uluer, et al. Impact of CFTR Modulation on Intestinal pH, Motility, and Clinical Outcomes in Patients With Cystic Fibrosis and the G551D Mutation. Clin Transl Gastroenterol 2017; 8: e81. [PMID: 28300821].
- Flume PA, Liou TG, Borowitz DS, Li H, Yen K, Ordoñez CL, et al. Ivacaftor in subjects with cystic fibrosis who are homozygous for the F508del-CFTR mutation. Chest 2012; 142:718-724. [PMID: 22383668].
- Clancy JP, Rowe SM, Accurso FJ, Aitken ML, Amin RS, Ashlock MA, et al. Results of a phase IIa study of VX-809, an investigational CFTR corrector compound, in subjects with cystic fibrosis homozygous for the F508del-CFTR mutation. Thorax 2012; 67:12-8. [PMID: 21825083].
- Claire E Wainwright, J Stuart Elborn, Bonnie W Ramsey, Gautham Marigowda, Xiaohong Huang, Marco Cipolli, et al. Lumacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N Engl J Med 2015; 373: 220-231. [PMID: 25981758].
- Steven M Rowe, Cori Daines, Felix C Ringshausen, Eitan Kerem, John Wilson, Elizabeth Tullis, et al. Tezacaftor-Ivacaftor in Residual-Function Heterozygotes with Cystic Fibrosis. N Engl J Med 2017; 377:2024-2035. [PMID: 29099333].
- Jennifer L Taylor-Cousar, Anne Munck, Edward F McKone, Cornelis K van der Ent, Alexander Moeller, Christopher Simard, et al. Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del. N Engl J Med 2017; 377:2013-2023. [PMID: 29099344].
- Davies JC, Moskowitz SM, Brown C, Horsley A, Mall MA, McKone EF, et al. VX-659-Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis and One or Two Phe508del Alleles. N Engl J Med 2018; 379:1599-1611. [PMID: 30334693].
- Nauman Chaudary. Triplet CFTR modulators: future prospects for treatment of cystic fibrosis. Ther Clin Risk Manag 2018; 14:2375-2383. [PMID: 30584312].
- Keating D, Marigowda G, Burr L, Daines C, Mall MA, McKone EF, et al. VX-445-Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis and One or Two Phe508del Alleles. N Engl J Med 2018; 379:1612-1620.
- Cystic Fibrosis Foundation Patient Registry. 2017 annual data report to the center directors. Bethesda (MD): Cystic Fibrosis Foundation; 2018.
- Pavel Drevinek, Tacjana Pressler, Marco Cipolli, Kris De Boeck, Carsten Schwarz, Florilene Bouisset. Antisense oligonucleotide eluforsen is safe and improves respiratory symptoms in F508DEL cystic fibrosis. J Cyst Fibros 2020; 19:99-107. [PMID: 31182369].
- M W Konstan, D R VanDevanter, S M Rowe, M Wilschanski, E Kerem, I Sermet-Gaudelus, et al. Efficacy and safety of ataluren in patients with nonsense-mutation cystic fibrosis not receiving chronic inhaled aminoglycosides: The international, randomized, double-blind, placebo-controlled Ataluren Confirmatory Trial in Cystic Fibrosis (ACT CF). J Cyst Fibros 2020; 19:595-601. [PMID: 31983658].
- Sriram Vaidyanathan, Ameen A Salahudeen, Zachary M Sellers, Dawn T Bravo, Shannon S Choi, Arpit Batish, et al. High-efficiency, selection-free gene repair in airway stem cells from cystic fibrosis patients rescues CFTR function in differentiated epithelia. Cell Stem Cell 2020; 26:161-171. [PMID: 31839569].
- Nicholas E King, Shingo Suzuki, Cristina Barillà, Finn J Hawkins, Scott H Randell, Susan D Reynolds, et al. Correction of airway stem cells: genome editing approaches for the treatment of cystic fibrosis. Hum Gene Ther 2020; 31:956-972. [PMID: 32741223].
- Kristin P Colling, Melena D Bellin, Sarah J Schwarzenberg, Louise Berry, Joshua J Wilhelm, Ty Dunn, et al. Total pancreatectomy with intraportal islet autotransplantation as a treatment of chronic pancreatitis in patients with cftr mutations. Pancreas 2018; 47:238-244. [PMID: 29206667].
- Chirag S Desai, Jennifer S Vonderau, Xiaobo Ma, Marilyn Hanson, Xiumin Xu, Aisha Khan. The first report of total pancreatectomy and islet cell autotransplantation for pancreatic cystosis in patient with cystic fibrosis. Pancreas 2019; 48:e54-e55. [PMID: 31206471].



