Keywords
Carcinoma, Adenosquamous; Pancreas; Sequence Analysis, DNA
Abbreviations
ASCOP Adenosquamous carcinoma of the pancreas;
MSKCC Memorial Sloan Kettering Cancer Center; MSK-IMPACT
Memorial Sloan Kettering - Integrated Mutation Profiling of Actionable
Cancer Targets; PDAC Pancreas ductal adenocarcinoma
INTRODUCTION
Pancreatic ductal adenocarcinoma (PDAC) is the
fourth most common cause of cancer related death
with an estimated 53,070 cases occurring in the United
States in 2016 [1]. The most common histological
subtype of exocrine pancreas cancer is adenocarcinoma.
Adenosquamous carcinoma of the pancreas (ASCOP)
represents an independent entity seen in 0.4-4% of all
exocrine pancreatic cancer cases with distinct clinical and
histological features when compared to pancreatic ductal
adenocarcinoma (PDAC) [2, 3, 4, 5]. Histologically, ASCOP
is characterized by the presence of both adenomatous glandular differentiation and keratinizing squamous
carcinoma constituents, with a 30% squamous carcinoma
component in coexistence with ductal adenocarcinoma
required to establish an ASCOP diagnosis [6, 7, 8]. The
etiological development of ASCOP remains inconclusive
with a number of different theories proposed [9].
Patients with ASCOP have a clear unmet need given
a poor prognosis and no defined standard treatments
other than those extrapolated from PDAC. ASCOP is
associated with an aggressive clinical course with poorer
clinical outcomes as compared to PDAC with an increased
tendency to have a larger tumor size, nodal positivity,
vascular invasion or perineural invasion and be poorly
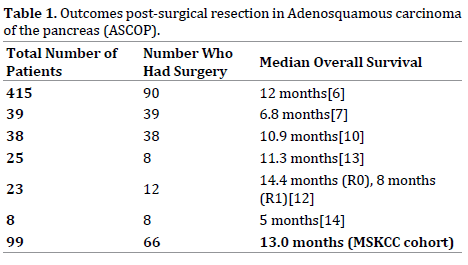
differentiated [6, 7, 10, 11]. Despite surgical resection for some patients with ASCOP, survival outcomes are poor
and range from 5 to 14.4 months [6, 7, 10, 12, 13, 14], see Table 1. For patients with metastatic disease, survival is
dismal with a typical survival outcomes of three to four
months reported [6, 11, 15].
Although progress has been in made in the molecular
characterization of PDAC this has not yet translated
into significant improvements in clinical outcome. Prior
attempts have also been made to perform molecular
profiling in ASCOP. In early reports KRAS, TP53 and SMAD4 alterations were observed [13, 16, 17, 18]. In a recent
analysis, mutations in the UPF1 gene were identified in 18
out of 23 ASCOP patients [19, 20]. UPF1 encodes an RNA
helicase involved in the nonsense-mediated RNA decay
(NMD) pathway which selectively degrades mRNAs that
harbor premature nonsense codons. Another recent study
using a number of molecular techniques depicted the
molecular profile of 23 patients with ASCOP identifying
KRAS alterations in all patients profiled with other rarer
alterations observed [21]. Recent genomic analyses of
pancreas cancer have described four molecular subtypes
with MYC activation, TGF-β (transforming growth factor
– beta) signaling, TP53 mutations, TP63 upregulation,
hypermethylation and autophagy associated with a
squamous subtype [22]. The amplification of MYC has also
been observed in ASCOP by whole-exome sequencing [23].
To further delineate both the clinical landscape
and molecular findings in ASCOP we collected
clinicopathological data on a cohort of 99 patients with
ASCOP evaluated at MSKCC and used an in-house exon
capture assay (Integrated Mutation Profiling of Actionable
Cancer Targets, or MSK-IMPACT) to interrogate the
mutation and copy number status of 410 oncogenes and
tumor suppressor genes commonly altered in cancer within
a subset cohort of 16 patients with available tissue [24]. In
our analysis we aimed to describe the clinical outcomes for
patients with this disease and identify potential actionable
targets within these tumors.
METHODS
Samples
Patients diagnosed with ASCOP from January 2000 to
July 2015 were identified from a prospectively maintained
database following IRB approval. Formalin fixed paraffin
(FFPE) samples and matched blood identified for genomic
sequencing were also collected under an approved IRB
protocol. For all specimens, representative hematoxylin and
eosin slides were reviewed by a board-certified pancreas
pathologist to confirm adenosquamous diagnosis prior to
submission for genomic profiling and samples were
reviewed to determine the optimal component for
sequencing. DNA was extracted as previously described [24].
Target Capture and Sequencing
The MSK-IMPACT assay detects mutations and copy
number alterations using DNA derived from frozen and formalin-fixed, paraffin-embedded (FFPE) tissue [24].
This assay requires as little as 15 ng of input DNA and
has high specificity and sensitivity using both frozen and
FFPE as input. The assay has been designed to sequence all
coding exons of 410 cancer-associated genes and provide
98% power to detect mutations with a true variant allele
frequency of 10%, novel mutations down to a threshold
of 5% variant allele frequency and mutations at recurrent
hotspots down to a threshold of 2% variant allele frequency.
Prior validation experiments have demonstrated the
accuracy, reproducibility, and sensitivity of the MSKIMPACT
assay and formed the basis of the assay approval
as a clinical test by the New York State Department of
Health [24]. Tumor and germline DNA from each sample
was processed to generate bar-coded libraries which were
subjected to exon capture using custom-designed probes.
Read pairs were assigned to the corresponding tumor
samples according to each barcode identity and then
aligned to the reference human genome. Both tumor and
matching normal DNA were run simultaneously to identify
germline single nucleotide polymorphisms (SNPs). All
data was then visualized and manually curated using the
Integrated Genomics Viewer.
Sequence Analysis
All sequence data was processed with an extensively
developed and optimized sequence analysis pipeline. Read
alignment, quality, performance metrics, post-processing,
somatic mutation and DNA copy number alteration detection
and variant annotation were performed using standard
best practices, a suite of validated open-source methods,
and custom analytics. Reads were aligned to the NCBI
reference human genome assembly using the Burrows
Wheeler alignment tool and processed using Picard tools
and the Genome Analysis Toolkit (GATK) pipeline followed
by multiple sequence realignment [25, 26]. Somatic point
mutations and indels were detected with the MuTect and
SomaticIndelDetector algorithms, respectively, while copy
number alterations were defined by seqCBS [27].
Statistics
Prespecified genomic alterations included in our MSKIMPACT
assay were analyzed. For known oncogenes,
we included point mutations and amplifications. For
tumor suppressors, truncating mutations (nonsense,
frameshift indels) and deletions were analyzed. Survival
was studied using Kaplan–Meier test. Overall survival
was calculated from date of diagnosis to either date of last
follow up or death. Time to disease relapse post curative
surgery was recorded from date of curative surgery to
date of disease relapse either radiographically or biopsy
proven when performed. Time to progression (TTP) on
first line chemotherapy for patients with metastatic/
unresectable disease was calculated from date of initiation
of chemotherapy to documented disease progression.
RESULTS
Ninety-nine patients with ASCOP were identified
from January 2000 to July 2015, of which sixteen cases had sufficient tissue and consent for molecular analysis.
Patient demographics and tumor characteristics are shown
in Table 2. Median age at diagnosis was 66 years (21-
88) with a male predominance (60.6%). The majority of
patients had stage II (54.5%) disease at diagnosis followed
by stage IV (28.3%); stage III (6.1%) and stage I (4.0%).
Tumors had location preponderance to the head of the
pancreas (56.6%). Sixty six patients underwent curative
resection of which 12 (18%) remain without evidence of
disease after a median follow up of 22 months (1-132).
Of the 66 patients who had curative intent resection,
thirty-nine (59.1%) had positive lymph nodes at time of
surgical resection. The median recurrence free interval
post curative surgical resection was four months (1-51).
Forty-three patients had disease recurrence post surgery
with the liver the most commonly affected site in 74.4% of
cases, followed by lung (13.9%), peritoneal (11.6%) and
local only recurrence in 4.7%.

We reviewed adjuvant chemotherapy regimens
utilized in ASCOP. Forty patients of 66 resected; (61%)
received adjuvant therapy in our institution. With regards
to other patients; nine patients followed up locally,
nine (14%) patients had metastatic disease on first
radiographic imaging post surgery, three patients did
not receive therapy, two patients died postoperatively,
two received neoadjuvant therapy and one patient had
a prolonged hospital stay post surgery which precluded
adjuvant therapy. The most frequent adjuvant regimen
was gemcitabine based therapy in twenty two patients.
Fluropyrimidine based therapy was used in eight patients.
The combination of gemcitabine and fluoropyrimidine
(capecitabine in all cases) was employed in a separate
8 patients and two patients enrolled to a clinical
trial. The addition of a platinum agent to gemcitabine
or fluropyrimidine based regimen occurred in four
cases (4/40; 10%). Sixteen patients (40%) received
chemoradiation as part of their adjuvant therapy regimen.
Six patients had locally advanced stage III disease. One
patient achieved surgical resection following neoadjuvant
GTX (gemcitabine, docetaxel and capecitabine) followed
by capecitabine based chemoradiation. Pathology revealed
a ypT2N0M0 tumor, this patient developed a solitary
metastatic lung nodule 22 months post surgery which was
surgically resected. The patient remains without evidence
of disease on follow up; 34 months from diagnoses and 5
months post lung metastasectomy.
Median overall survival (OS) for resected patients in
our cohort was 13 months (95%CI 7.5-18.5), Figure 1.
The median OS for patients with stage IV disease was 6
months (95%CI 0-13.2) Figure 2. With regards to first line
metastatic systemic chemotherapy regimens; gemcitabine
based therapy was utilized in 28 patients, fluropyrimidine
based in 17 patients, gemcitabine plus fluropyrimidine
based therapy in 4 patients and other regimens in two
patients [platinum plus irinotecan (n=1) and docetaxel
(n=1)]. Of these patients, 22 (45%) received a platinum as
part of therapy. Median TTP on first line chemotherapy for patients with metastatic/unresectable disease was three
months (range 1-11). Six patients with metastatic disease
were treated with best supportive care alone, three had de
novo metastatic disease and three patients with disease
relapse. Two patients with metastatic disease declined
chemotherapy.
Figure 1: Median Overall Survival (OS) for Resected Patients.
Figure 2: Median Overall Survival (OS) for patients with Stage IV disease.
Genomic Alterations Seen in ASCOP by MSK-IMPACT
With the aim of assessing somatic genetic alterations
in ASCOP, we analyzed 16 patients with ASCOP who had
both available tissue and consent using a capture-based,
massively parallel, next-generation sequencing assay
(MSK-IMPACT). Of the 16 patients, 10 had available
matched normal blood for comparison with the tumor
sample. Of the 16 patient samples sequenced, 14 were
from the primary tumor site and two samples were from
metastases (liver; n=2), Table 3. This likely represents a
sampling bias given ASCOP diagnosis may be challenging
from FNA sampling as compared to primary surgical
specimens. The majority of somatic alterations seen were
single base substitutions including non synonymous
coding changes, nonsense mutations with the remainder
amplifications or deletions.

MSK-IMPACT identified a variety of somatic alterations, Figure 3a. KRAS mutations were seen in all 16 patient
samples (100%) similar to previous reports [14, 21, 28].
Fifteen of the KRAS alterations were at codon 12 with
one at codon 61 (Q61H). TP53 mutations were seen in
12/16 (75%) of samples. Molecular alterations within
SMAD4 (38%) and CDKN2A (38%) occurred along with
a variety of other copy number alterations, Figure 3b.
Within the MSK-IMPACT set of 16 patients, two presented
with stage IV disease and fourteen had stage II disease at
initial presentation (stage IIA; n=5, stage IIB; n=9), Figure
3a,b. Twelve of the fourteen patients who had primary
resection developed metastatic disease with alterations in KRAS (100%; 12/12), TP53 (83%; 10/12), SMAD4 (42%,
5/12), CDKN2A (42%, 5/12) most commonly identified
in these patients. All five patients with SMAD4 alterations
(missense; n=2, truncating mutations; n=3) detected
by MSK-IMPACT in the primary tumor post resection
developed metastatic disease with a median time to
disease relapse of 7.5 months (range 5-16) as well as all
patients with Mcl-1 amplification (n=5) who had a median
time to disease relapse of 6 months, (range 1-16). One
other patient with a SMAD4 truncating mutation had stage
IV disease at presentation, Figure 3a.
Figure 3a: Most Frequent Somatic Mutations identified in 16 patients sequenced by MSK-IMPACT.
Figure 3b:Most Frequent Copy Number alterations detected in 16 patients identified by MSK-IMPACT.
The Phosphoinositide 3-Kinase (PI3K) pathway was
altered in 25% samples (4/16) through alterations in
PIK3R1, PIK3R2, PIK3CA and PTEN loss. We identified a
PIK3R1 missense E683K mutation in one patient sample
while the PIK3R2 I556 missense mutation has not been
previously characterized. The PIK3CA mutation was
a known oncogenic helical domain E542K missense
mutation. Mutations in the FAT1 gene located on 4q35.2 were seen in 3 (19%) patient samples. FAT1 missense
mutations observed included an R2567H missense
mutation (n=1), a T3424M missense mutation (n=1)
and both an M2845I and V2582M missense mutation in
one patient. The R2567H mutation has been previously
reported in a lung adenocarcinoma [29]. Two patients had
TGFBR1 (Transforming Growth Factor, Beta Receptor 1) in
frame mutations resulting in TGFBR1 loss.
STK11 (LKB1) is a tumor suppressor gene for which
one of its targets is the tuberin protein which inhibits
mTOR1 [30]. One patient had an STK11 missense mutation,
F354L. This mutation was previously described in the
germline of a child who developed neuroblastoma and a
follicular variant of papillary thyroid cancer [31]. It also
was observed in the germline in one Finnish patient and as
a somatic mutation in Korean patients with left sided colon
cancer [32, 33]. In a previous study in patients with left
sided colon cancer the F354L variant was seen not to alter
LKB1 kinase activity and felt to have a non-cancer inducing
effect [33]. Two TSC1 missense mutations were identified;
one T360N missense mutation previously described and
putatively linked to autism spectrum disorder and felt not
to be pathogenic in the tuberous sclerosis database [34].
The functional significance of the TSC1 V407M is unknown.
One patient had an RNF43 truncation mutation.
We also identified some original alterations. One
patient had an FGFR1 amplification, not been previously
described in ASCOP. One patient had a U2AF1 S34F
missense mutation. U2AF1 mutations have been noted in
cancers such as AML, squamous head and neck carcinoma,
endometrial carcinoma, urothelial bladder cancer and
breast, lung and colon adenocarcinoma previously [35].
U2AF1 alterations occur in ~11% of myelodysplastic
syndromes causing aberrant gene splicing with the S34F
missense mutation the most frequent mutation [36]. This
mutation to our knowledge is the first time U2AF1 has
been described in ASCOP. 31% (5/16) of patients had Mcl-
1 (Myeloid cell leukemia-1) amplification. Mcl-1part of the
anti-apoptotic subclass of Bcl-2 proteins and modulated
by oncogenic pathways such as MAPK, mTOR and PI3K signaling has been described in pancreas adenocarcinoma
but to our knowledge not in ASCOP [37, 38, 39, 40].
DISCUSSION
Adenosquamous cancer of the pancreas is a relatively
rare but particularly virulent form of pancreas cancer.
From our analyses and other previous reports ASCOP is a
systemic disease underpinned by poor survival outcomes.
Despite surgical resection with curative intent a median
OS of 13 months is very poor even relative to PDAC [6, 7, 10, 12, 13, 14] (Table 1). The rate of lymph node positivity
at time of surgery was high (59.1%), similar to previously
reported rates of 51.4% [6] and 76% [10]. In our cohort of
66 patients who had resection, nine (13.6%) had evidence
of metastases at time of first radiographic assessment
postoperatively. Relapse free interval post surgical
resection in our cohort was short at 4 months (1-51)
with disease most commonly affecting the liver (74.4%).
These factors suggest that a neoadjuvant systemic therapy
approach may be more appropriate in ASCOP thereby selecting a more appropriate patient population to
undergo future major resection.
Currently, no standard therapy exists for ASCOP in
the neoadjuvant, adjuvant or metastatic setting. Adjuvant
chemotherapy in ASCOP has been associated with
an improved survival in some single institution nonrandomized
series [13, 41]. In contrast neither adjuvant
chemotherapy nor radiation improved outcome in another
analysis [5]. Given the lack of established therapy most
regimens are based on those used in PDAC. No survival
difference was seen between gemcitabine based, 5-FU
based or combination of gemcitabine and 5-FU adjuvant
therapy in a prior retrospective analysis [41]. In our sample
set, gemcitabine based therapy was most frequently
employed in the adjuvant setting, with a doublet of
gemcitabine and capecitabine employed in eight separate
patients and platinum doublet in four patients. The use of
platinum agents in the adjuvant setting has been associated
with an improved survival of 19.1 months as compared to 10.7 months in a non-platinum group (p=0.015) in a
single institution non-randomized retrospective analysis
with no difference in outcome observed between cisplatin
or oxaliplatin [41]. In our cohort, 10% of cases received
adjuvant platinum therapy (oxaliplatin n=1, cisplatin
n=3) in combination with fluropyrimidine or gemcitabine
based therapy. Notably, none of our patients sequenced
displayed mutations in BRCA1, BRCA2, or PALB2 genes
that otherwise infer platinum sensitivity. In a prior report,
adjuvant chemoradiation (CRT) provided a survival benefit
as compared to surgery alone with characteristics such as;
tumor size of ≥3 cm, presence of vascular or perineural
invasion, nodal metastases and a squamous component
≥30% benefiting from CRT [10]. Incorporation of CRT as
part of the adjuvant treatment regimen in our cohort of
patients post resection was 40%. Given the small numbers
(16 patients) and retrospective nature of our data definitive
conclusions are not possible. Prospective trials with multi
institutional collaboration are required to definitely
corroborate the role of platinum and radiotherapy in the
adjuvant setting.
For those with metastatic disease prognosis was dismal
with a median OS of 6 months (95%CI 0-13.2) observed.
Gemcitabine based therapy was most frequent, with a
platinum agent utilized in 45% of patients who received
systemic chemotherapy. This rate was higher than our
10% platinum rate in the adjuvant setting. Nevertheless, median time to progression (TTP) was three months (1-
11) underscoring the inherent need to develop improved
therapeutic strategies for this aggressive disease. Putatively
this may be possible by understanding the molecular and
genomic drivers of this disease with the hope to develop
novel therapeutic agents.
MSK-IMPACT results for the 16 ASCOP tumors
sequenced revealed alterations in TP53, KRAS, SMAD4
and CDKN2A, similar to those found in PDAC. KRAS
mutations were seen in 100% of our samples which is
similar to previous reports [14, 16, 21]. Given the high
prevalence of KRAS mutations this highlights the need to
develop improved strategies to identify the Achilles heal
of mutated RAS. We did identify a number of potentially
actionable targets as well as novel alterations which may
represent susceptibility for therapeutic manipulation.
25% of patients had PI3K pathway alterations. Single
agent PIK3CA inhibition has minimal single agent activity
in pancreas cancer and given the cooccurrence of KRAS in
all cases, single agent PIK3CA inhibition is likely to have
a negligible therapeutic effect. Therefore combination
strategies with either chemotherapy or MEK inhibitors
ostensibly are needed. Of note, Weiss et al described an
ASCOP patient with both a KRAS and PIK3CA alteration
identified by next generation sequencing and a combination
of a PI3K inhibitor and MEK inhibitor was commenced with
a transient response [42]. In addition PIK3R1 mutations may be a potential target and are under assessment in
endometrial cancer [43]. Other potential amenable targets
include CDKN2A loss which can be potentially targeted with
CDK4/6 or MDM2 inhibitors [44]. One patient had RNF43
loss which in vitro is sensitive to porcupine inhibitors
[45]. Novel alterations in JAK3 were seen (19%) with in vitro
activity observed with the JAK3 inhibitor CP-690550 in ALL
with the V722I mutation, the same mutation identified in one
of our samples [46]. FAT1 mutations were observed in 19%
of cases and are linked with functional consequences upon
Wnt pathway dysregulation and activation [47]. Additionally,
one FGFR1 amplification not previously described in ASCOP
may be amenable to FGFR inhibitor therapy. Mcl-1 altered in
31% of cases has been tested in vivo and in vitro in pancreas
cancer and are targets which may benefit a subset of patients
[48]. This could be an important target since all five patients
with Mcl-1 amplification by MSK-IMPACT in their primary
tumor developed metastatic disease post resection.
As previously stated genomic analyses has characterized
a squamous type within four molecular subtypes of
pancreas cancer with characteristic signaling networks
including TP53 mutations and MYC activation [22]. By
MSK-IMPACT, 75% had TP53 mutations and one patient
had MYC amplification. MYC amplification is associated
with a poor outcome in ASCOP [23]. Our patient with MYC
amplification developed metastatic disease four months
post surgery and had a survival time of four months from
development of metastatic disease. Two patients both
never smokers with a head of pancreas primary location
had TGFBR1 in frame mutations resulting in TGFBR1 loss
both, an alteration described in other squamous cancers of
the head and neck and cervix [49, 50]. One patient had a
KDM6A mutation in our cohort which along with TP53 has
been associated with a pancreas squamous subtype [22].
In comparison to another previous study of molecular
profiling in ASCOP our rates of TP53, SMAD4, PIK3CA, and
EGFR alterations were higher with identical KRAS (100%),
BRAF (0%) and NRAS (0%) frequencies [21]. In addition,
we noted that all patients with SMAD4 alterations and Mcl-
1 amplification developed metastatic disease post surgery,
although numbers are small both SMAD4 and Mcl-1 have
previously been implicated as possible prognostic factors
[51, 52].
The etiology of ASCOP development is uncertain [9].
Our molecular analysis identified a GNAS alteration in one
patient in a known hotspot (R201H) occurring with a KRAS
mutation suggesting a highly MAPK dysregulated tumor.
GNAS alterations are associated with tumors that arise from
a precursor intraductal pancreatic neoplasm (IPMN) and are
felt to be specific for this event [53]. The presence of a GNAS
mutation in this case may infer an IPMN precursor lesion,
a rare event which has been previously described [54]. In
addition adenosquamous carcinoma has also been described
to occur in a mucinous cystadenoma [17].
In conclusion, our sequencing platform identified
a molecular signature for frequent alterations within
commonly affected PDAC genes (KRAS, TP53, SMAD4 and CDKN2A); however novel genes in U2AF1, PIK3R1 and
copy number alterations within Mcl-1 and FGFR1 were
identified. ASCOP is associated with poor outcomes despite
curative resection with a high rate of nodal positivity,
short interval to disease relapse and poor survival despite
surgery observed. Furthermore outcomes for metastatic
disease are underwhelming, therefore ASCOP represents
a disease with a clear need for improvement. Future
efforts can hopefully bridge the gap between molecular
complexity and therapeutic actionability for ASCOP.
Acknowledgements
We gratefully acknowledge funding from the Herman
Bott Pancreatic Cancer Research Fund and the Suzanne
Cohn Simon Pancreatic Cancer Research Fund in support
of this project.
Conflict of interest
The authors have no potential conflict of interest.
References
- https://www.cancer.org/research/cancerfactsstatistics/cancerfactsfigures2016/.
- Morohoshi T, Held G, Klöppel G. Exocrine pancreatic tumours and their histological classification. A study based on 167 autopsy and 97 surgical cases. Histopathology 1983; 7:645-61. [PMID: 6313514]
- Simone CG, Zuluaga Toro T, Chan E, Feely MM, Trevino JG, George TJ Jr. Characteristics and outcomes of adenosquamous carcinoma of the pancreas. Gastrointest Cancer Res 2013; 6:75-9. [PMID: 23936547]
- Madura JA, Jarman BT, Doherty MG, Yum MN, Howard TJ. Adenosquamous carcinoma of the pancreas. Arch Surg 1999; 134:599-603. [PMID: 10367867]
- Katz MH, Taylor TH, Al-Refaie WB, Hanna MH, Imagawa DK, Anton-Culver H, Zell JA. Adenosquamous versus adenocarcinoma of the pancreas: a population-based outcomes analysis. J Gastrointest Surg 2011; 15:165-74. [PMID: 21082275]
- Boyd CA, Benarroch-Gampel J, Sheffield KM, Cooksley CD, Riall TS. 415 patients with adenosquamous carcinoma of the pancreas: a population-based analysis of prognosis and survival. J Surg Res 2012; 174:12-9. [PMID: 21816433]
- Okabayashi T, Hanazaki K. Surgical outcome of adenosquamous carcinoma of the pancreas. World J Gastroenterol 2008; 14:6765-70. [PMID: 19058301]
- Hruban RH, Klimstra DS. Tumors of the Pancreas. AFIP Atlas of Tumor Pathology. Washington, DC, 2007. Series 4, Fascicle 6 Washington, DC: American Registry of Pathology 177–181.
- Cihak RW, Kawashima T, Steer A. Adenoacanthoma (adenosquamous carcinoma) of the pancreas. Cancer 1972; 29:1133-40. [PMID: 5021607]
- Voong KR, Davison J, Pawlik TM, Uy MO, Hsu CC, Winter J, Hruban RH, et al. Resected pancreatic adenosquamous carcinoma: clinicopathologic review and evaluation of adjuvant chemotherapy and radiation in 38 patients. Hum Pathol 2010; 41:113-22. [PMID: 19801164]
- Imaoka H, Shimizu Y, Mizuno N, Hara K, Hijioka S, Tajika M, Kondo S, et al., Clinical characteristics of adenosquamous carcinoma of the pancreas: a matched case-control study. Pancreas 2014; 43:287-90. [PMID: 24518509]
- Smoot RL, Zhang L, Sebo TJ, Que FG. Adenosquamous carcinoma of the pancreas: a single-institution experience comparing resection and palliative care. J Am CollSurg 2008; 207:368-70. [PMID: 18722942]
- Kardon DE, Thompson LD, Przygodzki RM, Heffess CS. Adenosquamous carcinoma of the pancreas: a clinicopathologic series of 25 cases. Mod Pathol 2001; 14:443-51. [PMID: 11353055]
- Brody JR, Costantino CL, Potoczek M, Cozzitorto J, McCue P, Yeo CJ, Hruban RH, et al. Adenosquamous carcinoma of the pancreas harbors KRAS2, DPC4 and TP53 molecular alterations similar to pancreatic ductal adenocarcinoma. Mod Pathol 2009; 22:651-9. [PMID: 19270646]
- Olson MT, Siddiqui MT, Ali SZ. The differential diagnosis of squamous cells in pancreatic aspirates: from contamination to adenosquamous carcinoma. Acta Cytol 2013; 57:139-46. [PMID: 23406837]
- Murakami Y, Yokoyama T, Yokoyama Y, Kanehiro T, Uemura K, Sasaki M, Morifuji M, et al., Adenosquamous carcinoma of the pancreas: preoperative diagnosis and molecular alterations. J Gastroenterol 2003; 38:1171-5. [PMID: 14714256]
- Campman SC, Fajardo MA, Rippon MB, Kraegel SA, Ruebner BH. Adenosquamous carcinoma arising in a mucinous cystadenoma of the pancreas. J Surg Oncol 1997; 64:159-62. [PMID: 9047255]
- Ohtsubo K, Mouri H, Sakai J, Akasofu M, Yamaguchi Y, Watanabe H, Gabata T, et al., Pancreatic cancer associated with granulocyte-colony stimulating factor production confirmed by immunohistochemistry. J Clin Gastroenterol 1998; 27:357-60. [PMID: 9855271]
- Liu C, Karam R, Zhou Y, Su F, Ji Y, Li G, Xu G, et al., The UPF1 RNA surveillance gene is commonly mutated in pancreatic adenosquamous carcinoma. Nat Med 2014; 20:596-8. [PMID: 24859531]
- Chang YF, Imam JS, Wilkinson MF. The nonsense-mediated decay RNA surveillance pathway. Annu Rev Biochem 2007; 76:51-74. [PMID: 17352659]
- Borazanci E, Millis SZ, Korn R, Han H, Whatcott CJ, Gatalica Z, Barrett MT, et al., Adenosquamous carcinoma of the pancreas: Molecular characterization of 23 patients along with a literature review. World J Gastrointest Oncol 2015; 7:132-40. [PMID: 26380056]
- Bailey P, Chang DK, Nones K, Johns AL, Patch AM, Gingras MC, Miller DK, et al., Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016; 531:47-52. [PMID: 26909576]
- Witkiewicz AK, McMillan EA, Balaji U, Baek G, Lin WC, Mansour J, Mollaee M, et al., Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat Commun 2015; 6:6744. [PMID: 25855536]
- Cheng DT, Mitchell TN, Zehir A, Shah RH, Benayed R, Syed A, Chandramohan R, et al., Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A Hybridization Capture-Based Next-Generation Sequencing Clinical Assay for Solid Tumor Molecular Oncology. J Mol Diagn 2015; 17:251-64. [PMID: 25801821]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009; 25:1754-60. [PMID: 19451168]
- DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 2011; 43:491-8. [PMID: 21478889]
- Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, Gabriel S, et al., Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol 2013; 31:213-9. [PMID: 23396013]
- Smit VT, Boot AJ, Smits AM, Fleuren GJ, Cornelisse CJ, Bos JL. KRAS codon 12 mutations occur very frequently in pancreatic adenocarcinomas. Nucleic Acids Res 1988; 16:7773-82. [PMID: 3047672]
- https://bioinfo.mc.vanderbilt.edu/TSGene/gene_mutation.cgi?gene=2195.
- Sanchez-Cespedes M. A role for LKB1 gene in human cancer beyond the Peutz-Jeghers syndrome. Oncogene 2007; 26:7825-32. [PMID: 17599048]
- Buryk MA, Picarsic JL, Creary SE, Shaw PH, Simons JP, Deutsch M6, Monaco SE, et al., Identification of Unique, Heterozygous Germline Mutation, STK11 (p.F354L), in a Child with an Encapsulated Follicular Variant of Papillary Thyroid Carcinoma within Six Months of Completing Treatment for Neuroblastoma. Pediatr Dev Pathol 2015; 18:318-23. [PMID: ]
- Dong SM, Kim KM, Kim SY, Shin MS, Na EY, Lee SH, Park WS, et al., Frequent somatic mutations in serine/threonine kinase 11/Peutz-Jeghers syndrome gene in left-sided colon cancer. Cancer Res 1998; 58:3787-90. [PMID: 9731485]
- Launonen V, Avizienyte E, Loukola A, Laiho P, Salovaara R, Järvinen H, Mecklin JP, et al., No evidence of Peutz-Jeghers syndrome gene LKB1 involvement in left-sided colorectal carcinomas. Cancer Res 2000; 60:546-8. [PMID: 10676634]
- Bahl S, Chiang C, Beauchamp RL, Neale BM, Daly MJ, Gusella JF, Talkowski ME, et al., Lack of association of rare functional variants in TSC1/TSC2 genes with autism spectrum disorder. Mol Autism 2013; 4:5. [PMID: 23514105]
- Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, Xie M, et al. Mutational landscape and significance across 12 major cancer types. Nature 2013; 502:333-9. [PMID: 24132290]
- Shirai CL, Ley JN, White BS, Kim S, Tibbitts J, Shao J, et al. Mutant U2AF1 Expression Alters Hematopoiesis and Pre-mRNA Splicing In Vivo. Cancer Cell 2015; 27:631-43. [PMID: 25965570]
- Mills JR, Hippo Y, Robert F, Chen SM, Malina A, Lin CJ, Trojahn U, et al. mTORC1 promotes survival through translational control of Mcl-1. Proc Natl Acad Sci U S A, 2008; 105:10853-8. [PMID: 18664580]
- Ding Q, Huo L, Yang JY, Xia W, Wei Y, Liao Y, Chang CJ, et al., Down-regulation of myeloid cell leukemia-1 through inhibiting Erk/Pin 1 pathway by sorafenib facilitates chemosensitization in breast cancer. Cancer Res 2008; 68:6109-17. [PMID: 18676833]
- Kuo ML, Chuang SE, Lin MT, Yang SY. The involvement of PI 3-K/Akt-dependent up-regulation of Mcl-1 in the prevention of apoptosis of Hep3B cells by interleukin-6. Oncogene 2001; 20:677-85. [PMID: 11314001]
- Miyamoto Y, Hosotani R, Wada M, Lee JU, Koshiba T, Fujimoto K, Tsuji S, et al., Immunohistochemical analysis of Bcl-2, Bax, Bcl-X, and Mcl-1 expression in pancreatic cancers. Oncology 1999; 56:73-82. [PMID: 9885381]
- Wild AT, Dholakia AS, Fan KY, Kumar R, Moningi S, Rosati LM, Laheru DA, et al., Efficacy of platinum chemotherapy agents in the adjuvant setting for adenosquamous carcinoma of the pancreas. J Gastrointest Oncol 2015; 6:115-25. [PMID: 25830031]
- Weiss GJ, Liang WS, Demeure MJ, Kiefer JA, Hostetter G, Izatt T, Sinari S, et al. A pilot study using next-generation sequencing in advanced cancers: feasibility and challenges. PLoS One 2013; 8:e76438. [PMID: 24204627]
- Cheung LW, Hennessy BT, Li J, Yu S, Myers AP, Djordjevic B, Lu Y, et al., High frequency of PIK3R1 and PIK3R2 mutations in endometrial cancer elucidates a novel mechanism for regulation of PTEN protein stability. Cancer Discov 2011; 1:170-85. [PMID: 24204627]
- Eilers G, Czaplinski JT, Mayeda M, Bahri N, Tao D, Zhu M, Hornick JL, et al., CDKN2A/p16 Loss Implicates CDK4 as a Therapeutic Target in Imatinib-Resistant Dermatofibrosarcoma Protuberans. Mol Cancer Ther 2015; 14:1346-53. [PMID: 25852058]
- Jiang X, Hao HX, Growney JD, Woolfenden S, Bottiglio C, Ng N, Lu B, et al. Inactivating mutations of RNF43 confer Wnt dependency in pancreatic ductal adenocarcinoma. Proc Natl Acad Sci U S A 2013; 110:12649-54. [PMID: 23847203]
- Yin C, Sandoval C, Baeg GH. Identification of mutant alleles of JAK3 in pediatric patients with acute lymphoblastic leukemia. Leuk Lymphoma, 2015; 56:1502-6. [PMID: 25146434]
- Morris LG, Kaufman AM, Gong Y, Ramaswami D, Walsh LA, Turcan Ş, Eng S, et al. Recurrent somatic mutation of FAT1 in multiple human cancers leads to aberrant Wnt activation. Nat Genet 2013; 45:253-61. [PMID: 23354438]
- Abulwerdi F, Liao C, Liu M, Azmi AS, Aboukameel A, Mady AS, Gulappa T, et al., A novel small-molecule inhibitor of mcl-1 blocks pancreatic cancer growth in vitro and in vivo. Mol Cancer Ther 2014; 13:565-75. [PMID: 24019208]
- Paterson IC, Matthews JB, Huntley S, Robinson CM, Fahey M, Parkinson EK, Prime SS. Decreased expression of TGF-beta cell surface receptors during progression of human oral squamous cell carcinoma. J Pathol 2001; 193:458-67. [PMID: 11276004]
- Xu XC, Mitchell MF, Silva E, Jetten A, Lotan R. Decreased expression of retinoic acid receptors, transforming growth factor beta, involucrin, and cornifin in cervical intraepithelial neoplasia. Clin Cancer Res 1999; 5:1503-8. [PMID: 10389939]
- Yamada S, Fujii T, Shimoyama Y, Kanda M, Nakayama G, Sugimoto H, Koike M, et al., SMAD4 expression predicts local spread and treatment failure in resected pancreatic cancer. Pancreas 2015; 44:660-4. [PMID: 25760429]
- Shigemasa K, Katoh O, Shiroyama Y, Mihara S, Mukai K, Nagai N, Ohama K. Increased MCL-1 expression is associated with poor prognosis in ovarian carcinomas. Jpn J Cancer Res 2002; 93:542-50. [PMID: 12036450]
- Furukawa T, Kuboki Y, Tanji E, Yoshida S, Hatori T, Yamamoto M, Shibata N, et al. Whole-exome sequencing uncovers frequent GNAS mutations in intraductal papillary mucinous neoplasms of the pancreas. Sci Rep 2011; 1:161. [PMID: 22355676]
- Karasaki H, Kono T, Ono Y, Maejima T, Mizukami Y. Adenosquamous cell carcinoma derived from intraductal papillary mucinous neoplasm of the pancreas confirmed by genetic analysis. J Clin Oncol 33; 2015 (suppl 3; abstr 275).





