Introduction
Environmental diseases (ENVDs) are non-communicable diseases that result when people are chronically exposed to toxic environmental chemicals. Other contributory causes of ENVDs include radiation, pathogens, allergens and psychological stress. These other causative agents, however, are minor compared with the chemical ones, which are the primary focus of this review. ENVDs are generally late-onset, appearing only after numerous toxic exposures. In recent years, however, the age of onset has been trending lower [1]. For all the ENVDs discussed, it is appreciated that genetic as well as environmental factors contribute to the onset of these diseases. It has been shown, however, that incidences of ENVDs are greatly enhanced by exposures to toxic environmental chemicals, and that more than one exogenous agent may trigger any given disease. The World Health Organization estimates that as much as 24% of global disease is caused by environmental exposures [2]. It has also been shown that 40% of cancers world wide can be prevented by lifestyle choices [3].
Hundreds of diseases fall under the definition just offered [4]. It is beyond the scope of this writing to address them all. The ENVDs discussed here are those that have been extensively studied and written on, thus providing a basis for reaching reasonable scientifically certain conclusions. Table 1 contains a list of the ENVDs addressed here and representative references for these [5-15]. The reader is referred to these citations for additional reference material.
| Disease |
|
| Metabolic: |
Type 2 diabetes (T2D)
Metabolic syndrome (MET-S) |
| Cardiovascular: |
myocardial infarction
atherosclerosis
hypertension
coronary heart disease
peripheral heart disease
ischemic heart disease
cardiac autonomic function |
| Neurodevelopmental: |
autism spectrum disorder (ASD)
attention deficit hyperactivity disorder (ADHD) |
| Neurodegenerative: |
Alzheimer's disease (AD)
Parkinson's disease (PD)
Amyotrophic lateral sclerosis (ALS) |
| Neurologicalimpairments: |
cognitive effects
motor deficits
sensory deficits
peripheral nervous system effects |
| Immunological: |
Allergic responses including chemical sensitivity
systemic sensitization autoimmune diseases |
| Musculoskeletal: |
Rheumatoid arthritis osteoporosis |
| Respiratory: |
Asthma
chronic obstructive pulmonary disease (COPD) |
| Cancer: |
childhood leukemia
other childhood cancers
breast cancer
prostate cancer
kidney cancer
numerous other cancers |
Table 1: Environmental diseases associated with chronic exposures to lipophilic exogenous toxicants and references for these [5-15]. References are to review publications. The reader is referred to the numerous references contained in these sources for primary material.
All of the diseases in Table 1 have reached epidemic and pandemic proportions in the past two generations. The dramatic increase of environmental disease prevalence with time can be seen from plots of disease percent increases versus time, from the 1950's to the present time. Such plots for numerous diseases produce hyperbolic curves, with examples being autism and autism spectrum disorders [16], type 2 diabetes [17], and obesity [18] in the United States. The slopes of these curves exactly correspond to those of plots for chemical production and use versus time, as illustrated by data for synthetic chemical production [17], and increased pesticide use versus time, much of which is dictated by the increased use of genetically engineered crops and global warming [19,20]. Figure 1, which shows the increase in incidence of autism in the United States from 1975 to the present, is representative of these relationships. Other disease increase rates that follow this curve include; childhood cancers, onset of dementia, other neurological diseases, breast, prostate and numerous other cancers as well both male and female infertility [4]. World wide energy production from combustion of fossil fuel use and its resultant air and water pollution increases also follow the slope of the curve in Figure 1 [21].
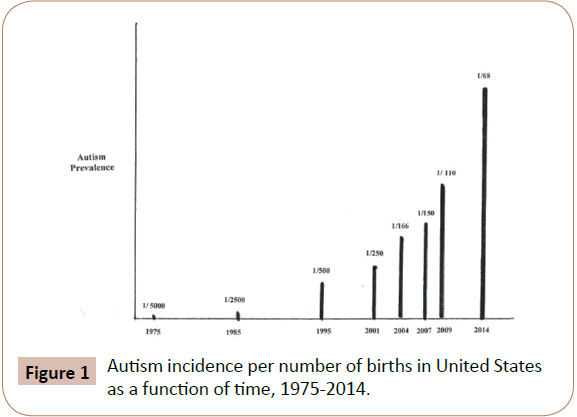
Figure 1: Autism incidence per number of births in United States as a function of time, 1975-2014.
The rapid increase in the incidence of ENVD occurred as a result of industrialization, changes in farming protocols and the increases in exposures to chemicals released into the environment as a result of these activities. Life style changes, including increased use of tobacco and the widespread introduction of processed foods also greatly contributed to the ENVD epidemics [4]. Before 1950, largely agrarian China was largely shielded from dramatic increases in non-communicable diseases. In recent decades, however, with rapid industrialization and resultant environmental impact, the Chinese people have experienced the same ENVD acceleration as those in the Western industrialized nations have, but over a much shorter period of time [22]. Environmental pollution, tobacco use and the obesity rate, for example, have dramatically increased [23]. Thus the Chinese "experiment" has served to highlight the cause and effect relationship between chemical exposure and environmental disease onset.
Chemical Toxicity
Historical
Acute exposures to high concentrations of toxic chemicals have been known for a long time to induce predictable deleterious health effects, but low concentrations of such chemicals have historically been believed to be benign to humans, In fact, regulatory agencies have assigned acceptable levels of exposure, known as permissible exposure levels (PELs) time weighted averages for 8 hours of exposure (TWAs) and no observed effect concentration values (NOECs) at which exposures presumably have no toxic effects [24].
Toxicity of Chemical Mixtures
Toxic effects of mixtures of chemicals with similar modes of action have been believed to be additive, while the effects of exposures to mixtures of chemicals with dissimilar toxic effects have been assumed to be benign as long as the concentrations of each was below established toxic levels. Synergistic and potentiated effects of dissimilar toxic chemicals have also been known for a long time. A review of the toxicological literature pre-2003 revealed that exposures to chemical mixtures had at times resulted in low concentration level toxicity, unpredicted target organ attack or greater than anticipated toxicity than from the known toxicology of individual chemical species in the mixtures. The studies that reported these unusual effects from exposures to chemical mixtures conceded that unknown factors were responsible for the observed effects [23].
Toxicity of Mixtures of Lipophilic and Hydrophilic Chemicals
Thus, health effects resulting from environmental exposures to toxic chemicals below regulatory agency established limits remained unexplained until 2003, when for the first time; it was shown that exposures to mixtures of chemicals containing at least one lipophilic and one hydrophilic species produced acute and chronic toxic effects even at very low level concentrations. In all of the chemical mixture exposure cases cited in the pre- 2003 literature and those reported for the first time in 2003 that described unusual and unpredicted toxic effects, the chemicals comprising the mixtures contained at least one lipophilic and hydrophilic component. It was hypothesized that the lipophilic component, which can penetrate mucous membranes, promoted the permeability of the hydrophilic species which would otherwise not permeate the lipophilic membranes. This hypothesis was supported by evidence from the literature in which it is shown that lipophilic chemicals are routinely added to hydrophilic pharmaceuticals that alone would not penetrate tissue at rates sufficient for clinical usefulness [25].
It was reported in 2003 that exposures to mixtures of lipophiles and hydrophiles produced enhanced toxicities at higher concentrations and, surprisingly, that such mixtures targeted organs and systems not known to be affected by the individual species and that different mixtures attacked different body organs and systems with each mixture acting as a unique toxic agent [25]. Though the original study focused on respiratory, central nervous system, liver, gastrointestinal and cardiovascular system effects, subsequent studies revealed that virtually every organ and system in the body is affected by low level exposures to mixtures of lipophilic and hydrophilic chemicals. Numerous examples, with no dissenting citations, were found in the literature to support these findings [24].
Disease Clusters
Cancer Clusters
The study of disease clusters affecting non-related individuals offers the opportunity to relate toxic effects of environmental chemicals with the genetic components factored out. In 2004, it was reported that that previously unexplained cancer clusters could be attributed to chronic exposures to mixtures of lipophiles and hydrophiles. A study based on an analysis of 12 cancer clusters that had previously been reported in the literature, and were not predicted from a consideration of the etiologies of the individual chemicals of exposure, was undertaken. It was shown that in each instance the cluster that ensued was attributed to exposures to mixtures of lipophilic and hydrophilic environmental chemicals. The cancer clusters included childhood leukemia, prostate cancer, testicular cancer, brain cancer and intracranial neoplasms in the children of mothers and fathers who were exposed and colorectal cancer [26].
Sequential Absorption
Sequential Absorption of Lipophiles
It has been well established that high molecular weight lipophilic exogenous chemicals such as polychlorinated biphenyls (PCBs) and organochlorine pesticides (OCs) are retained in the body for as long as decades [27-30]. In 2012 it was reported that even very low molecular weight lipophilic hydrocarbons, such as propane and butane, are retained for relatively long periods of time (4-7 days) and that constant exposure to these, via inhalation of polluted air, for example, can establish steady states of these compounds in the body [9]. It was further reported in that study and elsewhere [24] that due to the retention of lipophilic species in the body, absorption of both lipophile and hydrophile need not occur simultaneously. Rather, the absorption may be sequential, with the more slowly eliminated lipophiles absorbed initially followed by the subsequent absorption of hydrophiles to form the toxic mixtures. It is to be noted that hydrophilic chemicals are either not absorbed or are rapidly purged from the body in the absence of lipophilic species in which they are soluble [24].
Delayed Onset of Disease
The studies just discussed demonstrated that sequential absorption can account for the delayed onset of toxic consequences and led to the study of such induction of environmental disease. The first evidence for this was came in 2012, when it was reported that statin use by postmenopausal women resulted in an elevated risk for type 2 diabetes (T2D) [31]. An analysis of the data in that study revealed that all of the Statins which were reported on were lipophilic species and that the only Statin in clinical use at that time which did not raise the incidence of T2D was a hydrophilic species. It was hypothesized that though the lipophilic Statins are not as long lived in the body as PCBs and OCs, they are taken on a regular basis and establish a steady state in the body thus enabling them to act in a similar manner to other, longer retained, lipophilic chemicals [32].
Diabetes
In a study reported on in 2013, the association of lipophilic chemical exposure as a cause of type 2 diabetes (T2D) was examined [33]. The results of that study revealed that exposures to numerous lipophilic chemicals can lead to the increased incidence of T2D and that the slow onset of disease is due, at lease in part, to the initial absorption and the establishment of a steady state of lipophiles in body serum, followed by the sequential absorption of the hydrophiles as these become available. The sequential absorption of hydrophilic chemicals can occur minutes to years after the initial absorption of lipophilic species [33]. In T2D, dose-response relationships between serum levels of lipophiles and prevalence of disease have been reported [34,35]. Dose-response relationships have also been reported for neurological disease, cardiovascular disease, respiratory disease and cancer [36-40].
Cardiovascular and neurological diseases
In studies subsequent the diabetes study, it was reported that exposures to the identical lipophilic chemicals that were associated with increased incidence of T2D also triggered the onset of cardiovascular disease (CVD) [41], neurological disease; neurodevelopmental disease, including autism and autism spectrum disorders (ASD), as well as attention deficit hyperactivity disorder (ADHD); and neurodegenerative disease, including Alzheimer's disease (AD), Parkinson's disease (PD) and amyotrophic lateral sclerosis (ALS) [42]. Many other environmental diseases, that affect virtually all body systems, are also associated with exposure to and retention of exogenous lipophilic chemical species. These include: immunological [43], musculoskeletal [44,45] and respiratory diseases [46-48]; obesity [13] and numerous cancers [49-51]. Table 2 lists lipophilic exogenous chemicals, exposure to which has been shown to increase the incidences of these environmental diseases [13,33-51].
| Chemical |
Persistent organic pollutants:
polychlorinated biphenyls (PCBs)
organochlorine pesticides (OCs)
DDT
DDE
dieldrin
dioxins
furans
hexachlorobenzenes (HCBs)
polybrominated biphenyls (PBBs)
polybrominateddiphenyl ethers (PBDEs) |
Polynuclear aromatic hydrocarbons (PAHs)
acenaphthene
acenaphthylene
anthracine
benz[a]anthracine
benzo[a]pyrene
benzo[e]pyrene
benzo[b]fluoranthrene
benzo[g,h,i]perylene
benzo[j]fluroanthene
chrysene
dibenzo[a,h]anthracene
fluoanthene
fluorine
indeno[1,2,3-c,d]pyrene
naphthalene |
| C-10 to C-20 alkanes |
| Perfluorinated compounds |
Plastic formulation additives
Phthalates
diethylhexyl phthalate
dibutyl phthalate
di-n-pentyl phthalate
dicyclohexylphthalate
diallyl phthalate
diissodecyl phthalate
di-n-hexyl phthalate
diisobutyl phthalate
di-n-octyl phthalate
diisononyl phthalate
dihheptyl phthalate |
bisphenol-A
bisphenol-S |
| Phthalates |
Low molecular weight hydrocarbons (LMWHCs)
C-1 to C-8 alkanes
benzene
ethyl benzene
styrene
toluene
xylenes |
Halogenated alkanes and alkenes
vinyl chloride
trichloroethylene
tetrachloroethylene
methylene chloride |
Table 2: Lipophilic exogenous chemicals exposure to which has been shown to increase the incidence of numerous environmental diseases [13,33-51].
It has been reported that individuals morbid with one of the ENVDs in Table 1 also have high incidences of co-morbidities of other ENVDs [52]. In the United States alone, half of all adults have at least one environmental disease and more than a quarter of the adult population suffers from two or more co-morbid environmental diseases [53-55]. Accordingly, it is likely that an individual ill with one ENVD is likely to develop additional ENVD illness.
Co-morbidity of environmental diseases
All of the diseases listed in Table 1 are co-morbid with other environmental diseases that are known to be triggered by exposures to lipophilic chemicals [52]. Table 3 lists co-morbid disease pairs and references for each pair. Figure 2 [52] reprinted with permission) shows the co-morbidities of the 11 types of these diseases with each other. It is of note that of the 55 binary combinations possible, 45 (82%) % have been shown to be comorbid to date.
| Disease Pair |
| T2D - CVD |
| T2D - NRD |
| T2D - NDV |
| T2D - NDG |
| T2D - MSK |
| T2D - IMM |
| T2D - RES |
| T2D - CMS |
| T2D - OBS |
| T2D - CAN |
| CVD - NRD |
| CVD - NDV |
| CVD - NDG |
| CVD - MSK |
| CVD - IMM |
| CVD - RES |
| CVD - CMS |
| CVD - OBS |
| CVD - CAN |
| NRD - NDV |
| NRD - NDG |
| NRD - MSK |
| NRD - IMM |
| NRD - RES |
| NRD - CMS |
| NRD - OBS |
| NDV - RES |
| NDV - OBS |
| NDV - CAN |
| NDG - MSK |
| NDG - CAN |
| MSK - IMM |
| MSK - OBS |
| MSK - CAN |
| IMM - RES |
| IMM - CMS |
| RES - CMS |
| RES - OBS |
| RES - CAN |
| CMS - OBS |
| CMA - CAN |
| OBS - CAN |
Table 3: References for environmental disease co-morbidity pairs, for individual pair references see [52].
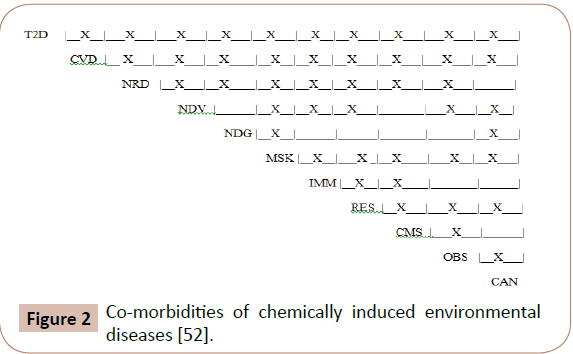
Figure 2: Co-morbidities of chemically induced environmental diseases [52].
Though different diseases involve attacks on widely disparate organs and systems, co-morbidity rates are high when individuals are exposed to environmental lipophilic toxins [9,52]. The onsets of co-morbid diseases do not follow set patterns. Published studies show that individuals with two co-morbid diseases, e.g., T2D and hypertension, are just as likely to become ill with one first as the other first [56]. The wide prevalence of co-morbid environmental diseases and the lack of a pattern of onset strongly suggest the common cause for these diseases that has been previously reported on [33,41,42] and that each lipophile: hydrophile pair acts as an independent toxicant [57].
Obesity
Obesity as an environmental disease
Obesity is an environmental disease that is a predictor of human adipose tissue concentration of POPs [58]. This is consistent with the fact that obesity is usually associated with CVS, T2D and other diseases, as adipose tissue releases the lipophiles it holds to the blood stream. Obesity is itself caused by POPs, phthalates, bisphenol A, polynuclear aromatic hydrocarbons (PAHs) and other exogenous lipophiles [13,59-63]. Being obese and having high serum lipophiles contributes to the absorption of these exogenous chemicals [58,64].
Obesity-lipophile-disease triangle
An Obesity - Lipophile - Disease triangle has been described as follows [52]. Exogenous lipophiles cause environmental diseases and obesity [52,65-70]. Environmental diseases (T2D, for example) cause the absorption of exogenous lipophiles and cause obesity [66,68,71]. Obesity causes environmental diseases and the absorption of lipophilic chemicals [72,73]. The Obesity- Lipophile- Disease triangle [52] is shown in figure 3.

Figure 3: Obesity-lipophile-disease triangle [52].
Absorption of Metals and other hydrophilic chemicals
Metals
The causes for the onset of the diseases listed in Table 1 are not limited to the action of lipophilic chemicals alone. Exposures to heavy metals and metalloids, which are listed in Table 4, have also been shown to increase the incidence of type 2 diabetes, cardiovascular diseases, neurological diseases and other ENVDs [74-86].
Hydrophilic transport
As described above, prior to 2015, is known that the toxic action of hydrophiles was based upon their solution in lipophiles and transport by lipophiles across the body's lipophilic membranes. Lipophilic species were defined as those with octanol: water partition coefficients (Kow) of 2.00 or greater and hydrophilic species were defined as those with Kow values of less than 2.00 [24,25]. It was empirically determined that optimal transport of hydrophiles by lipophiles across lipophilic membranes, and hence toxic effects of mixtures, were greatest when the Kow differences between hydrophiles and lipophiles was between 2.00 and 3.00 [25,57], and it was hypothesized that when Know differences were 5.00 or more, the solubility of hydrophile in lipophile was so low that no combined effect was observable [25]. Thus it was believed that metal ions, which are essentially insoluble in hydrocarbons, would act independently from hydrocarbons if exposure to both occurred simultaneously. As will be seen below, it has now been shown that metals are indeed transported across the body's lipophilic membranes by hydrocarbons when the hydrocarbons are planar aromatic species [87].
Metal transport
Some transition metals, iron and zinc, e.g., are required for body homeostasis. Yet, the onset of T2D, cardiovascular diseases, neurological diseases, neurodevelopmental diseases, neurodegenerative diseases, cancers and other ENVDs has been shown to be related to the action of elevated levels of Iron III ions (Fe+3) [73,88-94]. Until recently, it was mechanistically troubling as to how elevated levels of these metals permeate lipophilic cell membranes to enter body tissues. Recently, a physiochemical mechanism for ENVD onset based on the transport of metal ions through cell membranes via pi-bonding of metal ions to planar aromatic hydrocarbons was presented [87].
Cell Membranes
Membrane structure
Cell membranes are amphiphilic structures composed primarily of phospholipid bilayers with embedded proteins. The bilayers contain hydrophilic heads and hydrophobic (lipophilic) tails which contain embedded proteins. This arrangement allows the membrane to be selectively permeable to metal ions and other polar molecules, at the head, and through ion channels as well as the passive diffusion of lipophilic species through the tail. The lipid bilayer contains acyl groups that vary with cell type and include saturated and unsaturated fatty acid chains, with the two layers bonded by covalent disulfide bridges and van der Waals forces [95]. Two types of proteins are embedded in the lipid bilayer. The first, integral membrane proteins, interact primarily in the hydrophobic tails of cell membranes via nonpolar bonding. The second, peripheral proteins, interact primarily in the hydrophilic heads of cell membranes, via electrostatic and hydrogen bonds [96-99]. This variability allows particular cell membranes to selectively permit entry and egress of molecular species essential to their functioning. Thousands of different, unique membrane phospholipid structures have been identified. Each is characterized by the length and degree of saturation of its acyl chain, and all vary in membrane thickness, membrane fluidity, curvature and surface charge [98-100]. The differences in chemical composition and physical structure account for varying permeability’s of membranes.
Membrane permeability by lipophiles
Cell membranes are permeable to lipophilic exogenous chemicals. Such permeable lipophiles include but are not limited to: volatile and semi-volatile aliphatic hydrocarbons, single ring and polynuclear aromatic hydrocarbons (PAHs); phthalates; bisphenol-A; persistent organic pollutants such as polychlorinated biphenyls (PCBs), organochlorine pesticides (OCs) and polybrominated diphenyl ethers (PBDEs) fire retardants used in clothing; as well as food preservatives and disinfectants such as butylated hydroxytoluene (BHT), butylated hydroxyanisole (BHA), triclosan and parabens [24,25].
Effects of membrane permeation by lipophiles
Exogenous lipophilic chemicals (low molecular weight hydrocarbons, PAHs, pesticides and PCBs permeate through all cell membrane structures found in the human body, including the blood brain barrier (BBB) [101-106]. These chemicals penetrate into the hydrophobic tails of phospholipid cell membranes and accumulate there, leading to changes in membrane structure [107-109]. Such changes include: swelling of the membrane lipid bilayer, and thereby an increase in membrane surface area; and increase in membrane fluidity; inhibition of primary ion pumps; and an increase in proton and metal ion permeability [102-105]. These changes also bring about inhibition of embedded enzyme activity and the accumulation of exogenous lipophiles in the interior of the affected membranes [100,110].
Metal Toxicity
Metal absorption
As discussed above, Fe+3 is vital for cell homeostasis and is absorbed naturally via the polar heads and ion channels in healthy cells. Other vital transition metal ions are similarly absorbed. There are, however, two other routes by which Fe+3 and other transition metal ions may be absorbed through cell membranes. Firstly, cells with badly compromised lipid bilayers may cease to be barriers to hydrophilic species and permit direct passage of metal ions into the interior of cells [93,111]. Lipid bilayers can be compromised by the action of exogenous lipophiles or by surfactants that have polar ends which can bond to metal ions and non-polar ends which act as lipophiles and permeate cell membranes resulting in the transport of metal ions into cell interiors [115-117]. Secondly, cells that do not have previously damaged lipid bilayers may also be permeated by metal ions via pibonding of metals with planar aromatic molecular structures and co-passage of such metal/hydrocarbon complexes through the lipid bilayer. Pi-bonding of metal ions to aromatic ring structures is well established and the passage of metal-PAH complexes through lipophilic cell membranes has been previously reported [93]. Support for this hypothesis comes from the known toxic effects of mixtures of metals and aromatic hydrocarbons [118] as well as from consideration of the absorption of metal ions on the planar geometric shapes of PAHs, PCBs, phthalates, bisphenol-A, and mononuclear aromatics just described [93,114]. Metal ion/arene bonds arise from metal ion/pi-electron complexes which, due to the lipophilicity of the hydrocarbon component of the complex can permeate through the membrane lipid bilayer in a manner analogous to the permeation of lipophile/soluble hydrophile mixtures, as described above [24,25]. Following the penetration of the complex through the lipid bilayer, the complex dissociates, with the metal ions dissolving into the hydrophilic parts of the cell [93]. Both absorption routes lead to concentrations of metal ions that are toxic to cells.
Toxic consequences of metal absorption
Trivalent iron (Fe+3) can activate non-enzymatic blood coagulation resulting in the formation of parafibrin via a hydroxyl radical-catalyzed reaction that generates reactive oxygen species (ROS), which, in turn initiates oxidative stress [79,88,89]. Ferric ions cause the concomitant generation of hydroxyl radicals and polymerization of purified fibrinogen (FBG) [88]. In addition to iron, other transition metals have also been reported to generate hydroxyl radicals and thereby induce disease in humans. These metals, which include; silver, copper, vanadium, zinc, cobalt, mercury, lead, cadmium, arsenic, manganese and chromium are listed in Table 4 [74-78,80,89]. As is discussed below, production of ROS/RNS is the trigger of environmental disease.
Sequential absorption of metals
Essential metals are continually absorbed and desorbed into and out of healthy cells. Yet, as is the case with iron, excessive concentrations of these can lead to toxic consequences [88,89,90]. Non-essential metals such as cadmium, mercury, lead and chromium are toxic at very low concentration levels and have long half-lives in the human body with that of cadmium being on the order of 20-30 years. These transition metals are nonbiodegradable and can accumulate in the body with time [119]. The association between the sequential exposure to exogenous lipophilic chemicals and the onset of diabetes, cardiovascular disease, neurological disease, cancer and other diseases was discussed above [33,41,42]. Exposures to metals (including arsenic, cadmium, mercury lead, nickel, manganese, copper, zinc and chromium) are also known to induce all these diseases via the generation of ROS initiated oxidative stress [92-94,120-123]. Exposures to metals need not occur in one event and sequential exposures can, over time, generate toxic levels in the body [87]. It has also been reported that toxicity need not be due to a single metal ion species, but that total metal ion load may responsible for toxic effects [87]. This effect is complementary to the sequential absorption of lipophilic species and the reliance on total lipophilic load as indicative of prediction of permeability through lipophilic barriers and the onset of type 2 diabetes, cardiovascular disease, neurological diseases and co-morbidities [33,41,42,55]. Also, sequential absorption and accumulation in the body of both lipophiles and metal ions means that the body can be exposed to very low levels of both toxins and these toxins can accumulate in the body over time until critical levels are reached.
Total lipophilic and metal loads
It was also reported that both the lipophiles and the transition metal ions must be present in sufficiently high concentrations to induce disease onset [87]. At times, high concentrations of metal ions alone may be present and at times high concentrations of lipophiles alone may be present. Since all environmental exposures inevitably include absorption of metals and lipophilic chemicals, it is difficult to ascribe the onset of disease to metal or lipophile alone. Each alone, however, may not induce disease since the body continually metabolizes and/or eliminates both lipophiles and/or metals. Only when both species are present at critical levels, often involving sequential absorption, is disease triggered. It was additionally reported that total lipophilic load and total transition metal ion load, irrespective of the chemical nature of the individual species, are critical; since all lipophiles can permeate cell membrane lipophilic bilayers and numerous transition metal ions can pi-bond to a wide spectrum of planar aromatic hydrocarbons. Support for this dual chemical hypothesis comes from a consideration of the published health effects of both lipophilic chemicals and heavy metals. Both species have been shown to cause diabetes, cardiovascular disease, neurological disease (including neurological impairments, neurodevelopmental disorders and neurodegenerative diseases), immunological disease, obesity, cancer and act as endocrine disruptors. It is to be noted that although numerous studies have been published on the effects of a wide spectrum of individual lipophilic chemicals and heavy metals, only recently have the effects of mixtures of lipophiles and heavy metals been reported on [87]. Yet, it is essentially impossible for a human being living anywhere on the globe today not to be impacted by both lipophiles and transition metals at all times [124,125].
Body Protective Defenses
Metabolism and elimination
As discussed above, the human body naturally metabolizes and excretes exogenous toxic chemicals. Volatile and semi-volatile organic compounds are readily metabolized and eliminated. Though persistent organic pollutants, such as PCBs, PBBs, PBDEs, dioxins and furans are retained in adipose tissue for decades [27- 30], they slowly but continually partition into the blood stream from whence they can be absorbed by polyunsaturated oils, such as olive oil, and excreted [126-129]. Metals are also eliminated naturally via the action of natural chelating agents contained in spices, tea and other foods, for example [130-131]. In addition, the body has the ability to adapt to and repair damage caused by toxic chemicals [10]. These factors delay the onset of environmental disease. The liver and kidneys play vital roles in metabolism and elimination of toxic chemicals. A thorough discussion of these is, however, beyond the scope of this paper. The reader is referred to the literature for a fuller treatment of this subject.
Oxidative Stress
Oxidative stress can be induced in numerous ways. These include; exposure to exogenous toxic chemicals, including low molecular weight hydrocarbons, polynuclear aromatic hydrocarbons, chloro- and bromo- water disinfection by-products, phthalates, bis-phenol A, heavy metal ions; pharmaceuticals; exposure to radiation; as a result of environmental disease or infectious disease illness; psychological stress; sensory offensive agents; ingestion of certain foods and food additives; and obesity [6-8,25,55,87].
Chemical causes of Oxidative Stress
It is widely accepted that in vivo damage of biological molecules is initiated by reactive oxygen species (ROS) as well as by reactive nitrogen species (RNS) via oxidative stress (OS). It is believed and that elevated OS is responsible for a wide spectrum of diseases via molecular level toxic effects include lipid peroxidation, DNA attack, adduction, enzyme inhibition, oxidative attack on the central nervous system and cell signalizing, all of which have been linked to ENVDs including neurodegenerative diseases (Alzheimer's disease, Parkinson's disease, Amyotrophic Lateral Sclerosis), cancer, cardiovascular disease, diabetes, and others [132-135]. Electron transfer (ET) and the resultant formation of ROS can proceed via the reactivity of the metabolites of lipophilic molecules or via the action of metal ions to produce hydroxyl radicals [134,136].
Benzene serves as an example of a lipophilic molecule that undergoes a series of chemical reactions in the body to ultimately produce ROS. Benzene is metabolized to phenol, which is converted to catechol which is in turn oxidized to quinone [134]. Benzo[a]pyrene (BaP), a polynuclear aromatic hydrocarbon found in petroleum products and tobacco smoke and a known carcionogen, is another such example. It is the diol-epoxide of BaP, rather than BaP itself, that is the actual tumerogen [25].
Iron serves as an example of metal ion reactivity producing hydroxyl radicals. Molecular oxygen in the body can undergo a single electron reduction to form the superoxide radical anion, which in turn reacts with 2 protons to produce hydrogen peroxide (H2O2). H2O2 then reacts with divalent iron (Fe+2) to produce trivalent iron (Fe+3) and hydroxyl radical (OH*) via the Fenton reaction. Fe+3 may be converted back to Fe+2 via reaction with superoxide radical anion via the Haber-Weiss reaction. The net result is reaction of superoxide with hydrogen peroxide to produce hydroxyl radical [134].
All of the diseases listed in Table 1 as well as genitourinary, skin, hematological diseases and endocrine disorders including male and female infertility [137-149] have been associated with the generation of ROS. In addition, OS has also been shown to induce epigenetic effects [150-154].
The chemicals known to induce ROS are those listed in Tables 2 and 4. These include lipophilic species [155-160] and transition metal ions [161-164]. In addition, it has been reported that other chemical species, fructose for example, also induce oxidative stress [165].
| Arsenic |
| Cadmium |
| Chromium |
| Cobalt |
| Copper |
| Iron |
| Lead |
| Manganese |
| Mercury |
| Nickel |
| Silver |
| Tungsten |
| Vanadium |
| Zinc |
Table 4 Transition metal ions known to induce environmental disease [74-81].
Radiation
The parts of the electromagnetic spectrum (EMR) that induce oxidative stress include; ionizing, ultraviolet (UV), microwave and radiofrequency frequencies. It is well known that ionizing radiation causes oxidative stress [150]. UV radiation [166-168], microwave radiation [169-171] and radiofrequency radiation [172- 175] have also been associated with OS.
Biological stimuli
Biological stimuli that induce OS include; endogenous enzymes [176,177] and phagocytes [178,179], plant and animal allergens [180-183], and pathogens including bacteria, viruses and fungi [184-189].
Psychological stress
Chronic psychological stress (CPS) results in a prolonged state of oxidative stress, impairs the body's immune system response to anti-inflammatory signals [190] and results in the elevated secretion of glucocorticoids, a biomarker for which is cortisol [191,192]. CPS has been associated with increased risk of many diseases. These include; cardiovascular disease, diabetes, respiratory infections, autoimmune diseases, neurological diseases, including depression and psychiatric disorders, musculoskeletal disease, gastrointestinal and endocrine disorders, obesity and accelerated aging [192-199].
Aging
A discussion of OS as it relates to ENVDs requires attention to the part played by aging in the production of ROS. A widely held theory holds that, as part of aging, OS within mitochondria damages the mitochondria, which in turn leads to the production of increased quantities of ROS which cause further damage. Once it starts, this cycle leads to further damage and corresponding aging [200]. Oxidative stress has also been associated with the acceleration of telomere shortening and accelerated aging [201]. It is beyond the scope of this paper to fully explore the subject of aging. Numerous papers have been written on this subject, with the following representative of these [202-203].
Combined effects
The combination of CPS and stress induced by exogenous chemical exposure has been shown to produce additive OS effects [204-206]. These points out the need to consider all sources of stress - chemical, radiation, biological, aging as well as the social and psychological environment when evaluating total oxidative stress.
Environmental Disease Prevention
Absorption and ROS/RNS formation
As discussed above, two distinct events must occur for exogenous environmental chemicals to trigger disease: absorption and ROS/ RNS production. Prevention of ENVD prevalence can be reduced by limiting these events. That said, preventing chemically induced environmental disease, completely, is impossible, given the omnipresence of toxic chemicals everywhere on the globe. There are, however, steps that can be taken to significantly reduce the incidences of ENVDs. Proactive measures to prevent disease have been elucidated and are reviewed here. It is to be noted, however, that the preventative steps that are presented here cannot always be accomplished.
Individual actions
Exposure limiting steps that can be undertaken include those the individual can act on and those that society must take. One can control one's own lifestyle by addressing diet; immediate environment (home and work); avoiding the use of toxic chemical-containing products, avoiding the use of pesticides, avoiding the use of tobacco and tobacco smoke and secondary exposure to smoke, avoiding foods and personal care products that contain phthalates, bisphenol A and lipophilic preservatives such as triclosan, butylated hydroxy anisole (BHA), butylated hydroxy toluene (BHT) and parabens and limiting, to the extent that is medically advisable, the use of lipophilic pharmaceuticals. The individual can also choose to restrict the use of plastics containing phthalates and bisphenol A.
Societal actions
Steps that society can take include; educational programs and regulatory control of tobacco use, pesticides, persistent organic pollutants, plastics that exude phthalates and BPA and foods and personal care products that contain toxic components such as preservatives and solvents [25,207,208]. The development of green, non-polluting energy can greatly reduce toxic exposures by eliminating the release of lipophilic hydrocarbons, mercury, cadmium and other metals as well as by preventing global warming. Warmer environmental temperatures enhance the volatilization of POPs, pesticides and other organics into the air, require increased use of pesticides in farming, increase the rates of chemical reactions that lead to higher levels of ozone and secondary air pollutants and increase the risk of wildfires which spew large quantities of polluting species into the air [209,210].
Limitations to prevention
The measures to prevent ENVDs just discussed cannot always be implemented. Ignorance, socioeconomic status, lifestyle, peer pressure and economic interests of chemical manufacturers and polluters act counteractively to prevent the limiting of chemical exposures and ROS and RNS formation. There are also conflicting situations where well meaning people on both sides of an issue can reasonably disagree. Two examples of such situations serve to illustrate this point. DDT and its metabolite DDE are persistent organic pollutants that are causative of the ENVDs in Table 1. DDT, however, is still being used in parts of the world to control mosquitoes that carry malaria and other infectious diseases [211,212]. Water is routinely disinfected with chlorine and thus spares millions of people from exposures to water borne pathogens. Yet, the chlorinated and brominated hydrocarbon, haloketone and haloacetic acid byproducts of chlorination are human toxins [213,214]. These examples illustrate the often difficult choices that need to be made with regard to the use and subsequent human exposures to toxic chemicals.
Mediterranean diet
Reduction of ROS and RNS formation in the body can be readily accomplished through diet adherence to the Mediterranean diet, so named because it is the diet adhered to by populations bordering the Mediterranean Sea. This diet is rich in antioxidant phytochemicals, including phenolics, alkaloids, nitrogencontaining compounds, organosulfur compounds, phytosterols and carotenoids. More than 5,000 dietary phytochemicals have been identified in fruits, vegetables, whole grains, legumes, red wine, nuts and vegetable oils. An excellent review of the sources and benefits of dietary phytochemicals is presented by Liu [215]. In addition to the consumption of foods containing phytochemicals, the Mediterranean diet includes an increase in fish consumption, rich in antioxidant omega fatty acids, and a reduction of red meat consumption, a major source of serum triglycerides in which exogenous lipophiles accumulate.
Olive oil, and particularly extra virgin olive oil (EVOO), which is rich in phenolics, is the most representative food of the Mediterranean diet. Tree nuts are another major component of the diet. Both have been shown to reduce inflammation, oxidative stress and lower the risk of developing many ENVDs. These diseases include; Alzheimer's disease, Parkinson's disease, hypertension, diabetes, coronary heart disease and other cardiovascular diseases, obesity various cancers, anxiety and other central nervous system disorders [215-236].
Other preventive measures
The long onset times for ENVDs present opportunities to detoxify and otherwise protect the body of absorbed lipophiles and metals. Sauna has been demonstrated to reduce body levels of POPs and other lipophilic species and to lead to a relief of disease symptoms [237-239]. Detoxification by isolating patients in non-polluting environmental chambers along with diet control has been shown to result in reduced blood levels of volatile organic hydrocarbons as well as in the relief of disease symptoms [10,240]. Heavy metals can be removed via chelation by dietary components. Vitamins B1, B6, C and E have been shown to chelate heavy metals, as have garlic, ginger, onion, green tea, curry, tomatoes and other foods [241]. Selenium is an essential metal in the human body. When ingested in its selenite oxidation state, it has been shown to protect against metal induced neurotoxicity, renal toxicity and various cancers [242-244].
Lipophilic toxicity
Aromatic versus aliphatic compounds
The discussion presented here points out the need to avoid exposures to aromatic hydrocarbon-based lipophiles. It is well established that aromatic hydrocarbons are more toxic than aliphatic hydrocarbons of equal carbon number [245-247]. This may be seen from a comparison of blood: air partition coefficient (PC) and permissible exposure level (PEL) data. A higher PC indicates a greater propensity to absorb into blood and a lower PEL is indicative of greater toxicity. Table 5 demonstrates this by comparing PC and PEL data for C6-C8 hydrocarbons [87], reprinted with permission]. As can be seen from the data, aromatic hydrocarbons are more readily absorbed into blood and are more toxic than aliphatic hydrocarbons of equal carbon numbers. In addition, aromatic species, unlike aliphatic ones, can pi-bond with transition metals and thus facilitate their absorption.
| Carbon number |
Compound |
PC |
PEL |
| 6 |
benzene |
7.8 |
1 |
| |
cyclohexane |
1.3 |
300 |
| 7 |
toluene |
15.6 |
300 |
| |
n-heptane |
1.9 |
500 |
| 8 |
p-xylene |
28.4 |
100 |
| |
n-octane |
3.1 |
500 |
Table 5 Blood: air partition coefficient (PC) and permissible exposure level (PEL) data (given in parts per million) for C6-C8 hydrocarbons. The aromatic compound is listed first in each carbon number pair [87].
It is has been hypothesized [87] that the greater toxicity of the aromatic compounds is due to two factors. Firstly, their higher solubility in blood, from which these compounds can partition into membrane lipid bilayers. Secondly, the transport of transition metal ions dissolved in blood into cell structures via metal: pi-bonding with the planar aromatic species, as described above. Transition metal ions are always present in blood. Iron and some other transition metal ions, as also described above, are essential and naturally absorbed by the body form healthy foods. Non-essential transition metal ions enter the blood stream via inhalation, ingestion and dermal absorption. Accordingly, when aromatic hydrocarbons are absorbed into the blood, they can complex with the transition metals via pi-bonds and the complexed species absorbed through cell membranes, as described above, with resultant higher toxicity than is observed for aliphatic compounds which do not complex with transition metals.
Predicting environmental disease onset
Symptoms
ENVDs are, to a great extent, silent illnesses, in that in many instances, they are devoid of symptoms in their early stages which would alert the individual and/or the physician of the need to take action. As was seen above, dose response relationships between lipophilic body loads as well as between transition metal body load are associated with ENVD onset. For example, serum levels of POPs, including PCBs and OCs, associated with T2D have demonstrated a dose-response relationship between serum concentrations of these and the onset of disease [34,35,248,249]. Safe serum levels of these toxins, however, are yet to be established. Adipose tissue POPs concentrations have also been associated with the onset of T2D in a dose-response relationship [250]. These data, however, do not predict at what levels these pollutants in blood and adipose tissue are safe. These data also do not take into account the blood concentrations of other exogenous lipophilies nor transition metal concentrations. As was seen above, the chemicals associated with the onset of ENVDs are ROS/RON producing species, as are radiation, pathogens, aging and psychological stress. Accordingly, it is hypothesized here that total OS in the body is a better predictor of the potential for the onset of ENVDs than concentrations of individual chemical species alone and that biomarkers for total ROS are expected to better predict a person's likelihood of the onset of illness.
Biomarkers
Numerous biomarkers have been identified as indicators of exogenous toxic chemical-induced oxidative stress and have been related to the onset of cardiovascular, respiratory, autoimmune, musculoskeletal, liver, kidney and gastrointestinal diseases, as well as to diabetes, obesity and nervous system disorders [160,251-264]. Table 6 lists some of these biomarkers. Research to identify a single or combination of several biomarkers that would accurately predict increased probabilities of disease onset is ongoing in molecular epidemiology, a discipline devoted to the application of biomarkers to epidemiological studies [254].
| Homocystine |
| Total glutatione |
| Oxidized low density protein |
| Conjugated dienes |
| White blood cell count |
| Cortisol |
| C-reactive protein |
| Fibrinogen |
| Malondialdehyde |
| Diminished glomerular filtration rate |
| Increased ceatinine |
| Elevated fasting glucose |
| Low LDL cholesterol |
| High LDL cholesterol |
| Urinary aromatic hydrocarbon metabolites |
| Urinary phthalate metabolites |
| Bisphenol A metabolites |
Table 6. Partial list of biomarkers those are indicative of oxidative stress [251-262].
Future Challenges
New disease causative agents
New chemical causes of oxidative stress are constantly arising. Examples are: rare earth exposure as a result of the widespread use of these elements in electronic components [265]; and nanoparticle exposures as new uses for these are found on an almost daily basis [266,267].
Pharmaceuticals
The dramatic increase in the use of pharmaceuticals, in no small part, due to the exponential growth of ENVD prevalence, in itself contributes to the increase of the incidence of ENVDs. Two examples illustrate this point. Statins are widely prescribed to protect against cardiovascular disease. However, the most commonly prescribed Statins are themselves lipophiles which have been associated with the increased onset of type 2 diabetes [268,269]. Second-generation antipsychotic drugs are widely prescribed for children with attention deficit hyperactivity disorder (ADHD). These lipophilic drugs have been associated with an increased incidence of metabolic syndrome in children who have taken Quetiapine and Risperidone [270].
Global warming
No discussion of OS would be complete without a consideration of the effects of global warming on it. It is beyond the scope of this review to thoroughly examine this topic. Following, however, are some of the OS implications of global warming, which is a major contributor to OS for several reasons. First: elevated temperatures increase the solubility of PAHs and other lipophiles in water and also increase the vaporization of these species and hence result in higher air concentrations [271]. Second: elevated environmental temperatures accelerate the absorption of toxicants through respiratory, cutaneous and gastrointestinal routes and hence increase toxicity [210,271-273]. Third; elevated temperatures increase thermal stress, impair heat stress response and delay unfolded protein recovery [274-276]. Finally, warmer temperatures increase PAH-containing smoke from wildfires, airborne pollen and mycotoxin-producing molds, all of which stimulate OS [209,277].
Summary
The onset of environmental disease requires the penetration of toxicants through lipophilic cell membranes. Exogenous lipophilic chemicals not only accomplish this step, but also facilitate absorption of hydrophiles which serve as solvents and carriers. Lipophiles which have flat aromatic structures with pi-electron clouds also act as carriers for transition metal ions which pi-bond to these species. Disease results from OS due to the action of ROS/ RNS formed via the action of absorbed hydrophiles, hydrophilic metabolites of lipophiles and/or metals. Late onset of ENVDs is due to the slow deterioration of the body's immune system with age and the need for a buildup of sufficient lipophilic, hydrophilic and/or metal ion loads to overwhelm the body's ability to metabolize and eliminate such exogenous chemicals and to repair cellular damage. Absorption of lipophiles and metals need not be one-time events. These can be absorbed sequentially over time to build critical load levels. Sequential absorption points out the need to severely limit exposures to exogenous toxicants and for the drastic lowering of permissible levels of exposure to these chemicals. Exposures to aromatic hydrocarbons, which can undergo metal to pi-cloud bonding, and hence are more toxic than aliphatics hydrocarbons, should be severely restricted.
OS, which is the ultimate cause of ENVD, results not only from toxic chemical exposure, but also of the action of pathogens, exposure to radiation, aging and psychological stress. These causes are additive and when addressing OS, the total of all of these causes require consideration.
Globally, human exposures to exogenous chemicals are growing at exponential rates. This is manifest by not only increased prevalence of ENVDs, but also by increasingly lower ages of onset for type 2diabetes, obesity and cardiovascular diseases among others.
There are steps that can be taken to lower the incidence of ENVDs, but complete prevention of ENVDs is impossible, given the wide spread distribution of the causative chemicals and genetic predispositions. Biomarkers that determine total OS in the body would provide predictive indicators of increased vulnerability to the onset of ENVD and enable individuals to act to reduce total OS and hence disease.
References
- Wright A, Charlesworth B, Rudan I, Carothers A, Campbell H (2003) A polygenic basis for late-onset disease. Trends Genet 19: 97-106.
- Almost a quarter of all disease caused by environmental exposure (2006) World Health Organization.
- Cancer Research UK. Life choices behind more than four in 10 cancers.
- Murray CH, Vos T, Lozano R, Naghavi M, Flaxman AD, et al.(2010) Disability-adjusted life years (DALYs) for 291 diseases and injuries in 21 regions, 1990-2010: a systematic analysis for the global burden of disease study. Lancet 380: 2197-2223.
- Carpenter DO (2008) Environmental contaminants as risk factors for developing diabetes. Rev Environ Health 23: 59-74.
- Zeliger Hl (2013) Lipophilic chemical exposure as a cause of type 2 diabetes (T2D). Rev Environ Health 28: 9-20.
- Zeliger Hl (2013) Lipophilic chemical exposure as a cause of cardiovascular disease. Interdiscip Toxicol 6: 55-62.
- Zeliger Hl (2013) Lipophilic chemicals as a cause of neurological impairments, neurodevelopmental disorders and neurodegenerative diseases. Interdiscip Toxicol 6: 101-108.
- Zeliger Hl, Pan Y, Rea WJ (2012) Predicting co-morbidities in chemically sensitive individuals from exhaled breath analysis. Interdiscip Toxicol 5: 123-126.
- Rea WJ (1992) Chemical Sensitivity. Lewis Publishers.
- Edwards CJ, Cooper C (2006) Early environmental factors and rheumatoid arthritis. Clin Exp Immunol 143: 1-5.
- Sobieszcza?ska M, Jonkisz J, Tabin M, Laszki-Szcz?chor K (2013) Osteoporosis: genetic determinants and relationship with cardiovascular disease. Adv Clin Exp Med 22: 119-124.
- Langer P, Ukropec J, Kocan A, Drobna B, Radikova Z, et al. (2014) Obesogenic and diabetogenic impact of high organochlorine levels (HCB, p,p'-DDE, PCBs) on inhabitants in the highly polluted Eastern Slovakia. Endocr Regul 48: 17-24.
- Jacobs MM, Massey RI, Tenney H, Harriman E (2014) Reducing the use of carcinogens: the Massachusetts experience. Rev Environ Health 29: 319-340.
- Cogliano VJ, Baan R, Straif K, Grosse Y, Lauby-Secretan B, et al. (2011) Preventable exposures associated with human cancers. J Natl Cancer Inst 103: 1827-1839.
- Autism data U.S. Centers for Disease Control, Atlanta, GA.
- Neel BA, Sargis RM (2011) The paradox of progress: environmental disruption of metabolism and the diabetes epidemic. Diabetes 60: 1838-1848.
- U.S. Centers for Disease Control, Atlanta, GA. Obesity data.
- Benbrook CM (2012) Impacts of genetically engineered crops on pesticide use in the U.S. - the first sixteen years. Environ Sci Europe 24: 24.
- Chen CC, McCarl BA (2001) An investigation of the relationship between pesticide usage and climate change. Climate Change 50: 475-87.
- U.S. Energy Information Administration. U.S. Annual energy outlook, 2014. May 7, 2014.
- Yang G, Kong L, Zhao W, Wan X, Zhai Y, et al. (2008) Emergence of chronic non-communicable diseases in China. Lancet 372: 1697-1705.
- Huang C, Yu H, Koplan JP (2014) Can China diminish its burden of non-communicable diseases and injuries by promoting health in its policies, practices, and incentives? Lancet 384: 783-792.
- Zeliger HI (2011) Human toxicology of chemical mixtures.2nd ed. Elsevier London.
- Zeliger HI (2003) Toxic effects of chemical mixtures. Arch Environ Health 58: 23-29.
- Zeliger HI (2004) Unexplained cancer clusters: common threads. Arch Environ Health 59: 172-176.
- Yu GW1, Laseter J, Mylander C (2011) Persistent organic pollutants in serum and several different fat compartments in humans. J Environ Public Health 2011: 417980.
- Gallo MV, Schell LM, DeCaprio AP, Jacobs A (2011) Levels of persistent organic pollutant and their predictors among young adults. Chemosphere 83: 1374-1382.
- Kim MJ, Marchand P, Henegar C, Antignac JP, Alili R, et al. (2011) Fate and complex pathogenic effects of dioxins and polychlorinated biphenyls in obese subjects before and after drastic weight loss. Environ Health Perspect 119: 377-83
- M?llerov? D1, Kopeck? J (2007) White adipose tissue: storage and effector site for environmental pollutants. Physiol Res 56: 375-381.
- Culver AI, Oxkwnw IS, Balasubramanian R, et al. (2012) Statin use and the risk of diabetes mellitus in postmenopausal women in the Women's Health Initiative. Arch Int Med; 172: 144-52.
- Zeliger H (2012) Statin use and risk of diabetes.Arch Intern Med 172: 896.
- Zeliger HI (2013) Lipophilic chemical exposure as a cause of type 2 diabetes (T2D). Rev Environ Health 28: 9-20.
- Lee DH, Lee IK, Song K, Steffes M, Toscano W, Baker BA, et al. (2006) A strong dose-response relationship between serum concentrations of persistent organic pollutants and diabetes. Diabetes Care 29: 1638-44.
- Carpenter DO (2008) Environmental contaminants as risk factors for developing diabetes. Rev Environ Health 23: 59-74.
- Ren A, Qiu X, Jin L, Ma J, Li Z, et al. (2011) Association of selected persistent organic pollutants in the placenta with the risk of neural tube defects. Proc Natl Acad Sci USA 108: 12770-12775.
- Smith CJ, Fischer TH, Sears SB. Environmental tobacco smoke, cardiovascular disease, and the nonlinear dose-response hypothesis. Toxicol Sci 2000; 54(2):462-72.
- Gorell JM, Rybicki BA, Johnson CC, Peterson EL. Smoking and Parkinson's disease: a dose-response relationship. Neurology 1999; 52(1):115.
- Michel O, Nagy AM, Schroeven M, Duchateau J, N?ve J, et al. (1997) Dose-response relationship to inhaled endotoxin in normal subjects. Am J Respir Crit Care Med 156: 1157-1164.
- Law MR, Morris JK, Watt HC, Wald NJ (1997) The dose-response relationship between cigarette consumption, biochemical markers and risk of lung cancer. Br J Cancer 75: 1690-1693.
- Zeliger HI (2013) Lipophilic chemical exposure as a cause of cardiovascular disease. Interdiscip Toxicol 6: 55-62.
- Zeliger HI (2013) Exposure to lipophilic chemicals as a cause of neurological impairments, neurodevelopmental disorders and neurodegenerative diseases. Interdiscip Toxicol 6: 103-10.
- Marrie RA, Hanwell H (2013) General health issues in multiple sclerosis: comorbidities, secondary conditions, and health behaviors. Continuum (Minneap Minn) 19: 1046-1057.
- Al-Bishri J, Attar SM, Bassuni N, Al-Yofaiey A, Qutbudden H, et al. (2013) Comorbidity profile among patients with rheumatoid arthritis and the impact on prescription trend. Clin Med Insights: Arthritis and Musculoskeletal Disorders; 6: 11-18.
- Struijs JN, Baan CA, Schellevis FG, Westert GP, van den Bos GA (2006) Comorbidity in patients with diabetes mellitus: impact on medical health care utilization. BMC Health Serv Res 6: 84.
- Cazzola M, Segreti A, Calzetta L, Rogliani P (2013) Comorbidities of asthma: current knowledge and future research needs. Curr Opin Pulm Med 19: 36-41.
- L?pez Varela MV, Montes de Oca M, Halbert R, Mui?o A, T?lamo C, et al. (2013) Comorbidities and health status in individuals with and without COPD in five Latin American cities: the PLATINO study. Arch Bronconeumol 49: 468-474.
- van der Molen T (2010) Co-morbidities of COPD in primary care: frequency, relation to COPD, and treatment consequences. Prim Care Respir J 19: 326-334.
- Habib SL, Rojna M (2013) Diabetes and risk of cancer. ISRN Oncol 2013: 583786.
- S?rensen HT (2013) Multimorbidity and cancer outcomes: a for more research. Clin Epidemiol 5: 1-2.
- van Baal PH, Engelfriet PM, Boshuizen HC, van de Kassteele B, Schellevis FG, et al. (2011) Co-occurrence of diabetes, myocardial infarction, stroke and cancer, quantifying age patterns in the Dutch population using health survey data. Population Health Metrics 9: 51.
- Zeliger HI (2014) Co-morditities of environmental diseases: A common cause. Interdiscip Toxicol 7: 117-122.
- Jakovljevi?? M, Ostoji?? L (2013) Comorbidity and multimorbidity in medicine today: challenges and opportunities for bringing separated branches of medicine closer to each other. Psychiatr Danub 25 Suppl 1: 18-28.
- van Oostrom SH, Picavet HS, van Gelder BM, Lemmens LC, Hoeymans N, et al. (2012) Multimorbidity and comorbidity in the Dutch population - data from general practices. BMC Public Health 12: 715.
- Bauer UE, Briss PA, Goodman RA, Bowman BA (2014) Prevention of chronic disease in the 21st century: elimination of the leading preventable causes of premature death and disability in the USA. Lancet 384: 45-52.
- Sowers JR, Epstein M, Frohlich ED (2001) Diabetes, hypertension, and cardiovascular disease: an update. Hypertension 37: 1053-1059.
- Zeliger HI (2011) Respiratory toxicity of chemical mixtures: Predicting low level responses. Proc. Int. Conf. Indoor Air Quality, Austin, TX.
- Vaclavik E, Tjonneland A, Stripp C, Overvad K, Philippe Weber J, et al. (2006) Organochlorines in Danish women: predictors of adipose tissue concentrations. Environ Res 100: 362-370.
- Scinicariello F, Buser MC (2014) Urinary polycyclic aromatic hydrocarbons and childhood obesity: NHANES (2001-2006). Environ Health Perspect 122: 299-303.
- Dirinck EL, Dirtu AC, Govindan M, Covaci A, Van Gaal LF, et al. (2014) Exposure to persistent organic pollutants: relationship with abnormal glucose metabolism and visceral adiposity. Diabetes Care 37: 1951-1958.
- Choi J, Eom J, Kim J, Lee S, Kim Y (2014) Association between some endocrine disrupting chemicals and childhood obesity in biological samples of young girls: a cross-sectional study. Environ Toxicol Pharmacol 38: 51-57.
- Lee DH, Porta M, Jacobs DR Jr, Vandenberg LN (2014) Chlorinated persistent organic pollutants, obesity, and type 2 diabetes. Endocr Rev 35: 557-601.
- Simmons AL, Schlezinger JJ, Corkey BE (2014) What Are We Putting in Our Food That Is Making Us Fat? Food Additives, Contaminants, and Other Putative Contributors to Obesity. Curr Obes Rep 3: 273-285.
- Wang RY, Needham LL (2007) Environmental chemicals: from the environment to food, to breast milk, to the infant. J Toxicol Environ Health B Crit Rev 10: 597-609.
- M?llerov? D, Kopeck? J (2007) White adipose tissue: storage and effector site for environmental pollutants. Physiol Res 56: 375-381.
- La Merrill M, Emond C, Kim MJ, Antignac JP, Le Bizec B, et al. (2013) Toxicological function of adipose tissue: focus on persistent organic pollutants. Environ Health Perspect 121: 162-169.
- Riu A, McCollum CW, Pinto CL, Grimaldi M, Hillenweck A, et al. (2014) Halogenated bisphenol-A analogs act as obesogens in zebrafish larvae (Danio rerio). Toxicol Sci 139: 48-58.
- Kelishadi R, Poursafa P, Jamshidi F (2013) Role of environmental chemicals in obesity: a systematic review on the current evidence. J Environ Public Health 2013: 896789.
- Hardy OT, Czech MP, Covera S. What causes the insulin resistance underlying obesity? Curr Olin Endocrinol Diabetes Obes 2012; 19: 871-87.
- Kahn BB, Flier JS (2000) Obesity and insulin resistance. J Clin Invest 106: 473-481.
- Blatherwick NR (1921) Observations on blood fat in diabetes. J Biol Chem 49: 193-99.
- de Ferranti S, Mozaffarian D (2008) The perfect storm: obesity, adipocyte dysfunction, and metabolic consequences. Clin Chem 54: 945-955.
- Guh DP, Zhang W, Bansback N, Amarsi Z, Birmingham CL, et al. (2009) The incidence of co-morbidities related to obesity and overweight: a systematic review and meta-analysis. BMC Public Health 9: 88.
- Hsu PC, Liu MY, Hsu CC, Chen LY, Guo YL (1997) Lead exposure causes generation of reactive oxygen species and functional impairment in rat sperm. Toxicology 122: 133-143.
- Valko M, Morris H, Cronin MT (2005) Metals, toxicity and oxidative stress. Curr Med Chem 12: 1161-1208.
- Adlard PA, Bush AI (2006) Metals and Alzheimer's disease. J Alzheimers Dis 10: 145-163.
- Park HJ, Kim JY, Kim J, Lee JH, Hahn JS, et al. (2009) Silver-ion mediated reactive oxygen species generation affecting bactericidal activity. Water Res 43: 1027-32.
- Wang BJ, Sheu HM, Guo YL, Lee YH, Lai CS, et al. (2010) Hexavalent chromium induced ROS formation, Akt, NF-kappaB, and MAPK activation, and TNF-alpha and IL-1alpha production in keratinocytes. Toxicol Lett 198: 216-224.
- Lipinski B, Pretorius E (2013) Iron-induced fibrin in cardiovascular disease. Curr Neurovasc Res 10: 269-274.
- Martinez-Finley EJ, Gavin CE, Aschner M, Gunter TE (2013) Manganese neurotoxicity and the role of reactive oxygen species. Free Radic Biol Med 62: 65-75.
- Xu H, Finkelstein DI, Adlard PA (2014) Interactions of metals and Apolipoprotein E in Alzheimer's disease. Front Aging Neurosci 6: 121.
- Afridi HI, Kazi TG, Talpur FN, Arain S, Arain SS, et al. (2014)Distribution of arsenic, cadmium, lead and nickel levels in biological samples of Pakistani hypertensive patients and control subjects. Clin Lab 60: 1309-18.
- Bando K, Takahashi H, Kinbara M, Tanaka Y, Kuroishi T, et al. (2014) Resin monomers act as adjuvants in Ni-induced allergic dermatitis in vivo. J Dent Res 93: 1101-1107.
- Mat?s JM, Segura JA, Alonso FJ, M?rquez J (2010) Roles of dioxins and heavy metals in cancer and neurological diseases using ROS-mediated mechanisms. Free Radic Biol Med 49: 1328-1341.
- Agarwal S, Zaman T, Tuzcu EM, Kapadia SR (2011) Heavy metals and cardiovascular disease: results from the National Health and Nutrition Examination Survey (NHANES) 1999-2006. Angiology 62: 422-429.
- Park SJ, Lee JH, Woo SJ, Kang SW, Park KH (2015) Epidemiologic Survey Committee of Korean Ophthalmologic Society Five heavy metallic elements and age-related macular degeneration: Korean National Health and Nutrition Examination Survey, 2008-2011. Ophthalmology 122: 129-137.
- Zeliger HI, Lipinski B (2015) Physiochemical basis of human degenerative disease. Interdisip Toxicol 8: 101-107.
- Lipinski B, Pretorius E (2012) Novel pathway of iron???induced blood coagulation: implications for diabetes mellitus and its complications. Pol Arch Med Wewn 122: 115-122.
- Lipinski B, Pretorius E (2013) the role of iron-induced fibrin in the pathogenesis of Alzheimer's disease and the protective role of magnesium. Front Hum Neurosci 7: 735.
- Lipinski B (2014) Cancer wars: significance of protein unfolding in cancer and its inhibition with natural amphiphilic substances. Front Oncol 4: 183.
- Lipinski B. (2010) Prostate cancer vaccines, fibrin and selenium: a conceptual review. Open Prost Cancer J 3: 69-73.
- Chen YW, Yang CY, Huang CF, Hung DZ, Leung YM, et al. (2009) Heavy metals, islet function and diabetes development. Islets 1: 169-176.
- Gauthier PT, Norwood WP, Prepas EE, Pyle GG (2014) Metal-PAH mixtures in the aquatic environment: a review of c o-toxic mechanisms leading to more-than-additive outcomes. Aquat Toxicol 154: 253-269.
- Sharma B, Singh S, Siddiqi NJ (2014) Biomedical implications of heavy metals induced imbalances in redox systems. Biomed Res Int 2014: 640754.
- Boggs JM (1987) Lipid intermolecular hydrogen bonding: influence on structural organization and membrane function. Biochim Biophys Acta 906: 353-404.
- Lee AG (2003) Lipid-protein interactions in biological membranes: a structural perspective. Biochim Biophys Acta 1612: 1-40.
- Lee AG (2004) How lipids affect the activities of integral membrane proteins. Biochim Biophys Acta 1666: 62-87.
- Cornell BA, Separovic F (1983) Membrane thickness and acyl chain length. Biochim Biophys Acta 733: 189-193.
- de Kroon AI, Rijken PJ, De Smet CH (2013) Checks and balances in membrane phospholipid class and acyl chain homeostasis, the yeast perspective. Prog Lipid Res 52: 374-394.
- Holthuis JC, Menon AK (2014) Lipid landscapes and pipelines in membrane homeostasis. Nature 510: 48-57.
- Patel MM, Goyal BR, Bhadada SV, Bhatt JS, Amin AF (2009) Getting into the brain: approaches to enhance brain drug delivery. CNS Drugs 23: 35-58.
- Pardridge WM (2012) Drug transport across the blood-brain barrier. J Cereb Blood Flow Metab 32: 1959-1972.
- Qiu C, Cheng S, Xia Y, Peng B, Tang Q, et al. (2011) Effecs of subchronic banzo(a)pyrene exposure on neurotransmitter receptor gene expression in the rat hippocampus related with spatial learning and memory change. Toxicology 289: 83-90.
- Calderon-Garcideuenas L, Solt AC, Henriquez-Roldan C, Torres-Jardon R, Nuse B, Herritt L, et al. (2008) Heavy metals , islet function and diabetes development. Islets 1: 169.
- Gupta A. (1999) Functional impairment of blod-brain barrier following pesticide exposure during early development of rats. Human Experiment Toxicol 18: 174-79.
- Seelbach M, Chen L, Powell A, Coi YJ, Zhang B, et al. (2010) Polychlorinated biphenyls disrupt blood-brain barrier integrity and promote brain metastasis formation. Environ Health Perspect 118: 479-84.
- Endo S, Escher B, Goss KU (2011) Capacities of membrane lipids to accumulate neutral organic chemicals. Environ Sci Technol 45: 5912-5921.
- Librando V, Sarpietro MG, Castelli F (2003) Role of lipophilic medium in the absorption of polycyclic aromatic compounds by biomembranes. Environ Toxicol Pharmacol 14: 25-32.
- McIntosh TJ, Simon SA, MacDonald RC (1980) The organization of n-alkanes in lipid bilayers. Biochim Biophys Acta 597: 445-463.
- Sikkema J, de Bont JA, Poolman B (1994) Interactions of cyclic hydrocarbons with biological membranes. J Biol Chem 269: 8022-8028.
- Sikkema J, de Bont JA, Poolman B (1995) Mechanisms of membrane toxicity of hydrocarbons. Microbiol Rev 59: 201-222.
- Korchowiec B, Corvis Y, Viitala T, Feidt C, Guiavarch Y, et al. (2008) Interfacial approach to polyaromatic hydrocarbon toxicity: phosphoglyceride and cholesterol monolayer response to phenantrene, anthracene, pyrene, chrysene, and benzo[a]pyrene. J Phys Chem B 112: 13518-13531.
- Liland NS, Simonsen AC, Duelund L, Torstensen BE, Berntssen MH, et al. (2014) Polyaromatic hydrocarbons do not disturb liquid-liquid phase coexistence, but increase the fluidity of model membranes. Chem Phys Lipids 184: 18-24.
- Evans DR, Fackler NLP, Xie Z, Rickard CEF, Boyd PDW, et al. (1999) pi-arene cation structure and bonding. Solvation versus ligand binding in iron (III) tetraphenylprophyrin complexes of benzene, toluene, p-xylene and [60]Fullerane. J Am Chem Soc 121: 8466-74.
- Nielsen GD, Nielsen JB, Andersen KE, Grandjean P (2000) Effects of industrial detergents on the barrier function of human skin. Int J Occup Environ Health 6: 138-142.
- Singh A, Turner A. (2009) Surfactant-induced mobilisation of trace metals from estuarine sediment: implications for contaminent bioaccessibility and remediation. Environ Pollut 157: 646-53.
- Masakorala K, Turner A, Brown MT (2008) Influence of synthetic surfactants on the uptake of Pd, Cd and Pb by the marine macroalga, Ulva lactuca. Environ Pollut 156: 897-904.
- Wang W, Lampi MA, Huang XD, Gerhardt K, Dixon DG, et al. (2009) Assessment of mixture toxicity of copper, cadmium, and phenanthrenequinone to the marine bacterium Vibrio fischeri. Environ Toxicol 24: 166-177.
- Barbier O, Jacquillet G, Tauc M, Cougnon M, Poujeol P (2005) Effect of heavy metals on, and handling by, the kidney. Nephron Physiol 99: p105-110.
- Tchounwou PB, Yedjou CG, Patlolla AK, Sutton DJ (2012) Heavy metal toxicity and the environment. EXS 101: 133-164.
- Rani A, Kumar A, Lal A, Pant M (2014) Cellular mechanisms of cadmium-induced toxicity: a review. Int J Environ Health Res 24: 378-399.
- Bowman AB, Kwakye GF, Herrero Hern?ndez E, Aschner M (2011) Role of manganese in neurodegenerative diseases. J Trace Elem Med Biol 25: 191-203.
- Chen P, Parmalee N, Aschner M (2014) Genetic factors and manganese-induced neurotoxicity. Front Genet 5: 265.
- Bushnik T, Haines D, Levallois P, Levesque J, Van Oostdam J, et al. (2010) Lead and bisphenol A concentrations in the Canadian population. Health Rep 21: 7-18. 1. Wright A, Charlesworth B, Rudan I, Carothers A, Campbell H (2003) A polygenic basis for late-onset disease. Trends Genet 19: 97-106.
- Perez-Vazquez FJ, Flores-Ramirez R, Ochoa-Martinez AC, Orta-Garcia ST, Hernandez-Castro B, et al. (2015) Concentrations of persistent organic pollutants (POPs) and heavy metals in soil from San Luis Potosi, Mexico. Environ Monit Assess 187: 4119.
- Milbrath MO, Wenger Y, Chang CW, Emond C, Garabrant D, et al. (2009) Apparent half-lives of dioxins, furans, and polychlorinated biphenyls as a function of age, body fat, smoking status, and breast-feeding. Environ Health Perspect 117: 417-425.
- Ritter R, Scheringer M, MacLeod M, Schenker U, Hungerbuhler K. (2009) A kulti-individual pharmacokinetic model framework for interpreting time trends of persistentchemicals in human populations: application to a postban situation. Environ Health Perspect 117: 1280-86.
- Hooper K, She J, Sharp M, Chow J, Hewell N, et al. (2007) Depuration of polybrominated diphenyl ethers (PBDEs) and polychlorinated biphenyls (PCBs) in breast milk from California first-time mothers. (primiparae). Environ Health Perspect 115: 1271-75.
- Schnare DW, Robinson PC (1986) Reduction of the human body burdens of hexachlorobenzene and polychlorinated biphenyls. IARC Sci Publ : 597-603.
- Mandel S, Amit T, Reznichenko L, Weinreb O, Youdim MB (2006) Green tea catechins as brain-permeable, natural iron chelators-antioxidants for the treatment of neurodegenerative disorders. Mol Nutr Food Res 50: 229-234.
- Allen K, Cornforth D (2009) Effect of chelating agents and spice-derived antioxidants on myoglobin oxidation in a lipid-free model system. J Food Sci 74: C375-379.
- Kovacic P, Somanathan R (2012) Redox processes in neurodegenerative disease involving reactive oxygen species. Curr Neuropharmacol 10: 289-302.
- Kovacic P, Pozos RS, Somanathan R, Shangari N, O'Brien PJ (2005) Mechanism of mitochondrial uncouplers, inhibitors, and toxins: focus on electron transfer, free radicals, and structure-activity relationships. Curr Med Chem 12: 2601-2623.
- Kovacic P, Jacintho JD (2001) Mechanisms of carcinogenesis: focus on oxidative stress and electron transfer. Curr Med Chem 8: 773-796.
- Kovacic P, Somanathan R. (2014) Nitroaromatic compounds: environmental toxicity, carcinogenicity, mutagenicity therapy and mechanism. J Appl Toxicol 34: 810-24.
- Lipinski B1 (2011) Hydroxyl radical and its scavengers in health and disease. Oxid Med Cell Longev 2011: 809696.
- Jacobs DR Jr, Andersen LF, Blomhoff R. (2007) Whole-grain consumption is associated with a reduced risk of noncardiovascular, noncancer death attributed to inflammatory diseases in the Iowa Woemne's Health Study. Am J Clin Nutr 85: 1606-14.
- Khan MS, Tabrez S, Priyadarshini M, Priyamvada S, Khan MM (2012) Targeting Parkinson's - tyrosine hydroxylase and oxidative stress as points of interventions. CNS Neurol Disord Drug Targets 11: 369-380.
- Domej W, Oettl K, Renner W (2014) Oxidative stress and free radicals in COPD--implications and relevance for treatment. Int J Chron Obstruct Pulmon Dis 9: 1207-1224.
- Uttara B, Singh AV, Zamboni P, Mahajan RT (2009) Oxidative stress and neurodegenerative diseases: a review of upstream and downstream antioxidant therapeutic options. Curr Neuropharmacol 7: 65-74.
- Rath E, Haller D (2011) Inflammation and cellular stress: a mechanistic link between immune-mediated and metabolically driven pathologies. Eur J Nutr 50: 219-233.
- Zhang PY, Xu X, Li XC (2014) Cardiovascular diseases: oxidative damage and antioxidant protection. Eur Rev Med Pharmacol Sci 18: 3091-3096.
- Houstis N, Rosen ED, Lander ES (2006) Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature 440: 944-948.
- Kovacic P, Jacintho JD (2001) Reproductive toxins: pervasive theme of oxidative stress and electron transfer. Curr Med Chem 8: 863-892.
- Aitken RJ, Baker MA (2006) Oxidative stress, sperm survival and fertility control. Mol Cell Endocrinol 250: 66-69.
- Smith R, Kaune H, Parodi D, Madariaga M, Morales I, et al. (2007) [Extent of sperm DNA damage in spermatozoa from men examined for infertility. Relationship with oxidative stress]. Rev Med Chil 135: 279-286.
- Gupta S, Ghulmiyyah J, Sharma R, Halabi J, Agarwal A (2014) Power of proteomics in linking oxidative stress and female infertility. Biomed Res Int 2014: 916212.
- Agarwal A, Gupta S, Sharma RK (2005) Role of oxidative stress in female reproduction. Reprod Biol Endocrinol 3: 28.
- M?n?zo Y, Entezami F, Lichtblau I, Cohen M, Belloc S, et al. (2012) [Oxidative stress and fertility: false evidence and bad recipes]. Gynecol Obstet Fertil 40: 787-796.
- Szumiel I (2015) Ionizing radiation-induced oxidative stress, epigenetic changes and genomic instability: the pivotal role of mitochondria. Int J Radiat Biol 91: 1-12.
- Szarc vel Szic K, Declerck K, Vidakovi?? M, Vanden Berghe W (2015) From inflammaging to healthy aging by dietary lifestyle choices: is epigenetics the key to personalized nutrition? Clin Epigenetics 7: 33.
- Galligan JJ, Rose KL, Beavers WN, Hill S, Tallman KA, et al. (2014) Stable histone adduction by 4-oxo-2-nonenal: a potential link between oxidative stress and epigenetics. J Am Chem Soc 136: 11864-11866.
- Assies J, Mocking RJ, Lok A, Ruhe HG, Pouwer F, et al. (2014) Effects of oxidative stress on fatty acid- and one-carbon-metabolism in psychiatric and cardivascular disease comorbidity. Acta Psychiatr Scand 130: 163-80.
- Joven J, Micol V, Segura-Carretero A, Alonso-Villaverde C, Men?ndez JA; Bioactive Food Components Platform (2014) Polyphenols and the modulation of gene expression pathways: can we eat our way out of the danger of chronic disease? Crit Rev Food Sci Nutr 54: 985-1001.
- Hennig B, Hammock BD, Slim R, Toborek M, Saraswathi V, et al. (2002) PCB-induced oxidative stress in endothelial cells: modulation by nutrients. Int J Hyg Environ Health 205: 95-102.
- Jaiswal PK, Srivastava S, Gupta J, Thakur IS. (2012) Dibenzofuran induces oxidative stress, disruption of trans-mitochondrial membrane potential and G1 arres in human hepatoma cell line. Toxicoll Lett 214: 137-44.
- Hong YC, Park EY, Park MS, Ko JA, Oh SY, et al. (2009) Community level exposure to chemicals and oxidative stress in adult population. Toxicol Lett 184: 139-144.
- Mathieu-Denoncourt J, Wallace SJ, de Solla SR, Langlois VS (2015) Plasticizer endocrine disruption: Highlighting developmental and reproductive effects in mammals and non-mammalian aquatic species. Gen Comp Endocrinol 219: 74-88.
- Babu S, Uppu S, Claville MO, Uppu RM (2013) Prooxidant actions of bisphenol A (BPA) phenoxyl radicals: implications to BPA-related oxidative stress and toxicity. Toxicol Mech Methods 23: 273-280.
- Kumar J, Lind PM, Salihovic S, van Bavel B, Lind L, et al. (2014) Influence of persistent organic pollutants on oxidative stress in population-based samples. Chemosphere 114: 303-09.
- Szebtmihalyi K, Blazovics A, Vinkler R. (2003) Free radical properties of metal complexes. Acta Biologica Szegediensis 47: 107-09.
- Jomova K, Valko M (2011) Advances in metal-induced oxidative stress and human disease. Toxicology 283: 65-87.
- Valko M, Rhodes CJ, Moncol J, Izakovic M, Mazur M (2006) Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem Biol Interact 160: 1-40.
- Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, et al. (2007) Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol 39: 44-84.
- DiNicolantonio JJ, O'Keefe JH, Lucan SC (2015) Added fructose: a principal driver of type 2 diabetes mellitus and its consequences. Mayo Clin Proc 90: 372-381.
- Klamt F, Dal-Pizzol F, Bernard EA, Moreira JC (2003) Enhanced UV-mediated free radical generation; DNA and mitochondrial damage caused by retinol supplementation. Photochem Photobiol Sci 2: 856-860.
- Lesser MP. (1996) Elevated temperatures and ultraviolet radiation cause oxidative stress and inhibit photosynthesis in symbiotic dinoflagettates. Limnol Oceanog 41: 271-83.
- Svbodova AR, Galandakova A, Sianska J, Dolzal D, Ulrichoa J Vostalova J. (2011) Acute exposure to solar simulated ultraviolet radiation affects oxidative stress-related biomarkers in skin, liver and blood or hairless mice. Biol Pharm Bull 34: 471-79.
- Desai NR, Kesari KK, Agarwal A (2009) Pathophysiology of cell phone radiation: oxidative stress and carcinogenesis with focus on male reproductive system. Reprod Biol Endocrinol 7: 114.
- Sokolovic D, Djindjic B, Nikolic J, Bjelakovic G, Pavlovic D, et al. (2008) Melatonin reduces oxidative stress induced by chronic exposure of microwave radiation from mobile phones in rat brain. J Radiat Res 49: 579-586.
- Megha K, Deshmukh PS, Banerjee BD, Tripathi AK, Abegaonkar MP (2012) Microwave radiation induced oxidative stress, cognitive impairment and inflammation in brain of Fischer rats. Indian J Exp Biol 50: 889-896.
- Avci B, Akar A, Bilgici B, Tun?el ?K (2012) Oxidative stress induced by 1.8 GHz radio frequency electromagnetic radiation and effects of garlic extract in rats. Int J Radiat Biol 88: 799-805.
- Esmekaya MA, Ozer C, Seyhan N (2011) 900 MHz pulse-modulated radiofrequency radiation induces oxidative stress on heart, lung, testis and liver tissues. Gen Physiol Biophys 30: 84-89.
- De Iuliis GN, Newey RJ, King BV, Aitken RJ (2009) Mobile phone radiation induces reactive oxygen species production and DNA damage in human spermatozoa in vitro. PLoS One 4: e6446.
- Tkalec M, Malari?? K, Pevalek-Kozlina B (2007) Exposure to radiofrequency radiation induces oxidative stress in duckweed Lemna minor L. Sci Total Environ 388: 78-89.
- Ameziane-El-Hassani R, Talbot M, de Souza Dos Santos MC, Al Ghuzlan A3, Hartl D4, et al. (2015) NADPH oxidase DUOX1 promotes long-term persistence of oxidative stress after an exposure to irradiation. Proc Natl Acad Sci U S A 112: 5051-5056.
- Ishii N, Fujii M, Hartman PS, Tsuda M, Yasuda K, et al. (1998) A mutation in succinate dehydrogenase cytochrome b causes oxidative stress and ageing in nematodes. Nature 394: 694-697.
- Babior BM (2000) Phagocytes and oxidative stress. Am J Med 109: 33-44.
- Elbim C1, Pillet S, Prevost MH, Preira A, Girard PM, et al. (2001) The role of phagocytes in HIV-related oxidative stress. J Clin Virol 20: 99-109.
- Ritsick DR, Lambeth JD (2005) Spring brings breezes, wheezes, and pollen oxidases. J Clin Invest 115: 2067-2069.
- Brussino L, Badiu I, Sciascia S, Bugiani M, Heffler E, Guida G, et al. (2010) Oxidative stress and airway inflammation after allergen challenge evaluated by exhaled breath condensate analysis. Clin Exp Allergy 40: 1642-47.
- Basci A, Dharajiya N, Choudhury BK, Sur S, Boldogh I. (2005) Effect of pollen-mediated oxidative stress on immediate hypersensitivity reactions and late-phase inflammation in allergic conjunctivitis. J Allergy Clin Immunol 116: 836-43.
- Cho YS, Moon HB (2010) The role of oxidative stress in the pathogenesis of asthma. Allergy Asthma Immunol Res 2: 183-187.
- Lushchak VI (2011) Adaptive response to oxidative stress: Bacteria, fungi, plants and animals. Comp Biochem Physiol C Toxicol Pharmacol 153: 175-190.
- Grant SS, Hung DT (2013) Persistent bacterial infections, antibiotic tolerance, and the oxidative stress response. Virulence 4: 273-283.
- Ray A, Martinez BA, Berkowitz LA, Caldwell GA, Caldwell KA. (2014) mitochondrial dysfunction, oxidative stress, and neurodegeneration elicited by a bacterial metabolite in a C. elegans Parkinson's model. Cell Death Dis 5: 984.
- Beck MA, Handy J, Levander OA (2000) The role of oxidative stress in viral infections. Ann N Y Acad Sci 917: 906-912.
- Schwarz KB (1996) Oxidative stress during viral infection: a review. Free Radic Biol Med 21: 641-649.
- Dantas Ada S, Day A, Ikeh M, Kos I, Achan B, et al. (2015) Oxidative stress responses in the human fungal pathogen, Candida albicans. Biomolecules 5: 142-165.
- Miller GE, Cohen S, Ritchey AK (2002) Chronic psychological stress and the regulation of pro-inflammatory cytokines: a glucocorticoid-resistance model. Health Psychol 21: 531-541.
- Dhabhar FS (2014) Effects of stress on immune function: the good, the bad, and the beautiful. Immunol Res 58: 193-210.
- Aschbacher K, O'Donovan A, Wolkowitz OM, Dhabhar FS, Su Y, et al. (2013) Good stress, bad stress and oxidative stress: insights from anticipatory cortisol reactivity. Psychoneuroendocrinology 38: 1698-1708.
- Cohen S, Janicki-Deverts D, Doyle WJ, Miller GE, Frank E, Rabin BS, et al. (2012) chronic stress, glucocortioid receptor resistence, inflammation, and disease risk. PNAS 109: 5995-99.
- Costantini D, Marasco V, M?ller AP (2011) A meta-analysis of glucocorticoids as modulators of oxidative stress in vertebrates. J Comp Physiol B 181: 447-456.
- Aschenbacher K, Kornfeld S, Picard M, Puterman E, Havel PJ, et al.(2014) Chronic stress increases vulnerability to diet-related abdominal fat, oxidative stress, and metabolic risk. Psychoneuroendocrinology 46: 14-22.
- Maes M, Kubera M. Obuchowiczwa E, Goehler L, Brzeszcz J. (2011) Depression's multiple comorbidities explained by (neuro)inflammatory and oxidative & nitrosative stress pathways. Nuero Endocrinol Lett 32: 7-24.
- Halaris A (2013) Inflammation, heart disease, and depression. Curr Psychiatry Rep 15: 400.
- Halaris A (2013) Co-morbidity between cardiovascular pathology and depression: role of inflammation. Mod Trends Pharmacopsychiatri 28: 144-161.
- Salim S (2014) Oxidative stress and psychological disorders. Curr Neuropharmacol 12: 140-147.
- Romano AD, Serviddio G, de Matthaeis A, Bellanti F, Vendemiale G (2010) Oxidative stress and aging. J Nephrol 23 Suppl 15: S29-36.
- Crous-Bou M, Fung TT, Prescott J, Julin B, Du M, et al. (2014) Mediterranean diet and telomere length in Nurses' Health Study: population based cohort study. BMJ 349: g6674.
- Junqueira VB, Barros SB, Chan SS, Rodrigues L, Giavarotti L, et al. (2004) Aging and oxidative stress. Mol Aspects Med 25: 5-16.
- Andriollo-Sanchez M, Hininger-Favier I, Meunier N, Venneria E, O'Connor JM, et al. (2005) Age-related oxidative stress and antioxidant parameters in middle-aged and older European subjects: the ZENITH study. Eur J Clin Nutr 59 Suppl 2: S58-62.
- Abdel-Rahman A, Abou-Donia SM, El-Masry E, Shetty AK, Abou-Donia MB. (2004) Stress and combined exposure to low doses of pyridostigmine bromide, deet, and permethrin produce neurochemical and neuropathological alterations in cerebral cortex, hippocampus and cerebellum. J Toxicol Environ Health A 67: 163-92.
- Shepard JD, al'Absi M, Whitsett TL, Passey RB, Lovallo WR (2000) Additive pressor effects of caffeine and stress in male medical students at risk for hypertension. Am J Hypertens 13: 475-481.
- Gump BB, Reihman J, Stewart P, Lonky E, Granger DA, et al. (2009) Blood lead (Pb) levels: further evidence for an environmental mechanism explaining the association between socioeconomic status and psychophysiological dysregulation in children. Health Psychol 28: 614-20.
- Kushner RF, Sorensen KW (2013) Lifestyle medicine: the future of chronic disease management. Curr Opin Endocrinol Diabetes Obes 20: 389-395.
- Minich DM, Bland JS (2013) Personalized lifestyle medicine: relevance for nutrition and lifestyle recommendations. ScientificWorldJournal 2013: 129841.
- Kinney PL (2008) Climate change, air quality, and human health. Am J Prev Med 35: 459-467.
- Noyes PD, McElwee MK, Miller HD, Clark BW, Van Tiem LA, et al. (2009) The toxicology of climate change: environmental contaminants in a warming world. Environ Int 35: 971-986.
- Sahu SS, Gunasekaran K, Raju HK, Vanamail P, Pradhan MM, et al. (2014) Response of malaria vectors to conventional insecticides in the southern districts of Odisha State, India. Indian J Med Res 139: 294-300.
- Gunasekaran K, Sahu SS, Jambulingam P, Das PK (2005) DDT indoor residual spray, still an effective tool to control Anopheles fluviatilis-transmitted Plasmodium falciparum malaria in India. Trop Med Int Health 10: 160-168.
- Tardiff RG, Carson ML, Ginevan ME. (2006) Updated weight of evidence for an association between adverse reproductive and developmental effects and exposure to disinfection byproducts. Regul Toxicol Pharmacol 45: 185-205.
- Bove GE Jr, Rogerson PA, Vena JE (2007) Case control study of the geographic variability of exposure to disinfectant byproducts and risk for rectal cancer. Int J Health Geogr 6: 18.
- Liu RH (2013) Health-promoting components of fruits and vegetables in the diet. Adv Nutr 4: 384S-92S.
- Bouayed J, Rammal H, Soulimani R (2009) Oxidative stress and anxiety: relationship and cellular pathways. Oxid Med Cell Longev 2: 63-67.
- Hu N, Yu JT, Tan L, Wang YL, Sun L, et al. (2013) Nutrition and the risk of Alzheimer's disease. Biomed Res Int 2013: 524820.
- Mart?nez-Gonz?lez MA (2006) The SUN cohort study (Seguimiento University of Navarra). Public Health Nutr 9: 127-131.
- Martinez-Lapiscina EH, Clavero P, Toledo E, Estruch R, Salas-Salvado J, et al. (2012) Mediterranean diet improves cognition: the PREDIMED-NAVARRA randomised trial. J Neurol Neurosurg Psychiatry 84: 1318-1325.
- Sofi F, Cesari F, Abbate R, Gensini GF, Casini A (2008) Adherence to Mediterranean diet and health status: meta-analysis. BMJ 337: a1344.
- Estruch R, Ros E, Salas-Salvad? J, Covas MI, Corella D, et al. (2013) Primary prevention of cardiovascular disease with a Mediterranean diet. N Engl J Med 368: 1279-1290.
- L?pez-Miranda J, P?rez-Jim?nez F, Ros E, De Caterina R, Badim?n L, et al. (2010) Olive oil and health: summary of the II international conference on olive oil and health consensus report, Ja?n and C?rdoba (Spain) 2008. Nutr Metab Cardiovasc Dis 20: 284-294.
- Waterman E, Lockwood B (2007) Active components and clinical applications of olive oil. Altern Med Rev 12: 331-342.
- Cicerale S, Lucas LJ, Keast RS (2012) Antimicrobial, antioxidant and anti-inflammatory phenolic activities in extra virgin olive oil. Curr Opin Biotechnol 23: 129-135.
- Cicerale S, Conlan XA, Sinclair AJ, Keast RS (2009) Chemistry and health of olive oil phenolics. Crit Rev Food Sci Nutr 49: 218-236.
- Lucas L, Russell A, Keast R (2011) Molecular mechanisms of inflammation. Anti-inflammatory benefits of virgin olive oil and the phenolic compound oleocanthal. Curr Pharm Des 17: 754-768.
- Cicerale S, Lucas L, Keast R (2010) Biological activities of phenolic compounds present in virgin olive oil. Int J Mol Sci 11: 458-479.
- Gopinath B, Buyken AE, Flood VM, Empson M, Rochtchina E, et al. (2011) Consumption of polyunsaturated fatty acids, fish, and nuts and risk of inflammatory disease mortality. Am J Clin Nutr 93: 1073-1079.
- Banel DK, Hu FB (2009) Effects of walnut consumption on blood lipids and other cardiovascular risk factors: a meta-analysis and systematic review. Am J Clin Nutr 90: 56-63.
- Jiang R, Jacobs DR Jr, Mayer-Davis E, Szklo M, Herrington D, et al. (2006) Nut and seed consumption and inflammatory markers in the multi-ethnic study of atherosclerosis. Am J Epidemiol 163: 222-231.
- Jaceldo-Siegl K, Haddad E, Oda K, Fraser GE, Sabat? J (2014) Tree nuts are inversely associated with metabolic syndrome and obesity: the Adventist health study-2. PLoS One 9: e85133.
- Pan A, Sun Q, Manson JE, Willett WC, Hu FB (2013) Walnut consumption is associated with lower risk of type 2 diabetes in women. J Nutr 143: 512-518.
- Bao Y, Han J, Hu FB, Giovannucci EL, Stampfer MJ, et al. (2013) Association of nut consumption with total and cause-specific mortality. N Engl J Med 369: 2001-2011.
- Luu HN, Blot WJ, Xiang YB, Cai H, Hargreaves MK, et al. (2015) Prospective evaluation of the association of nut/peanut consumption with total and cause-specific mortality. JAMA Intern Med 175: 755-766.
- Tan SY, Dhillon J, Mattes RD (2014) A review of the effects of nuts on appetite, food intake, metabolism, and body weight. Am J Clin Nutr 100 Suppl 1: 412S-22S.
- Mattes RD, Kris-Etherton PM, Foster GD (2008) Impact of peanuts and tree nuts on body weight and healthy weight loss in adults. J Nutr 138: 1741S-1745S.
- Kilburn KH, Warsaw RH, Shields MG. (1989)Neurobehavioral dysfunction in firemen exposed to polychlorinated biphenyls (PCBs): possible improvement after detoxification. Arch Environ Health 44: 345-50.
- Krop J (1998) Chemical sensitivity after intoxication at work with solvents: response to sauna therapy. J Altern Complement Med 4: 77-86.
- Crinnion WJ (2011) Sauna as a valuable clinical tool for cardiovascular, autoimmune, toxicant- induced and other chronic health problems. Altern Med Rev 16: 215-225.
- Rea WJ, Pan Y. (1996) Reduction of chemical sensitivity by means of heat depuration, physical therapy and nutritional supplementation in a controlled environment. J Nutr Environ Med 6: 141-149.
- Zhai Q, Narbad A, Chen W (2015) Dietary strategies for the treatment of cadmium and lead toxicity. Nutrients 7: 552-571.
- Li WH1, Shi YC, Tseng IL, Liao VH (2013) Protective efficacy of selenite against lead-induced neurotoxicity in Caenorhabditis elegans. PLoS One 8: e62387.
- Ip C1 (1998) Lessons from basic research in selenium and cancer prevention. J Nutr 128: 1845-1854.
- Aslanturk A, Uzunhisarcikli M, Kalender S, Demir F (2014) Sodium selenite and vitamin E in preventing mercuric chloride induced renal toxicity in rats. Food Chem Toxicol 70: 185-190.
- Perbellini L, Brugnone F, Caretta D, Maranelli G (1985) Partition coefficients of some industrial aliphatic hydrocarbons (C5-C7) in blood and human tissues. Br J Ind Med 42: 162-167.
- Sato A, Nakajima T (1987) Pharmacokinetics of organic solvent vapors in relation to their toxicity. Scand J Work Environ Health 13: 81-93.
- Smith AQ, Campbell JL, Keys DA, Fisher JW (2005) Rat tissue and blood partition coefficients for n-alkanes (C8 to C12). Int J Toxicol 24: 35-41.
- Longnecker MP, Klebanoff MA, Brock JW, Zhou H; Collaborative Perinatal Project (CPP) (2001) Polychlorinated biphenyl serum levels in pregnant subjects with diabetes. Diabetes Care 24: 1099-1101.
- Wu H, Bertrand KA, Choi AL, Hu FB, Laden F, et al. (2013) Persistent organic pollutants and type 2 diabetes: a prospective analysis in the nurses' health study and meta-analysis. Environ Health Perspect 121: 153-161.
- Arrebola JP, Pumarega J, Gasull M, Fernandez MF, Martin-Olmedo P, et al. (2013) Adipose tissue concentrations of persistent organic pollutants and prevalence of type 2 diabetes in adults from Southern Spain. Environ Res 122: 31-37.
- Dalle-Donne I, Rossi R, Colombo R, Giustarini D, Milzani A (2006) Biomarkers of oxidative damage in human disease. Clin Chem 52: 601-623.
- Kumar J, Lind L, Salihovic S, van Bavel B, Ingelsson E, et al. (2014) Persistent organic pollutants and liver dysfunction biomarkers in a population-based human sample of men and women. Environ Res 134: 251-256.
- Ford ES, Cunningham TJ, Mannino DM (2015) Inflammatory markers and mortality among US adults with obstructive lung function. Respirology 20: 587-593.
- Ogino K, Wang DH (2007) Biomarkers of oxidative/nitrosative stress: an approach to disease prevention. Acta Med Okayama 61: 181-189.
- Pollack AZ, Mumford SL, Mendola P, Perkins NJ, Rotman Y, et al. (2015) Kidney biomarkers associated with blood, lead, mercury, and cadmium in premenopausal women: a prospective cohort study. J Toxicol Environ Health A 78: 119-31.
- Ettinger AS, Bovet P, Plange-Rhule J, Forrester TE, Lambert EV, et al. (2014) Distribution of metals exposure and associations with cardiometabolic risk factors in the "Modeling the Epidemiologic Transition Study". Environ Health 13: 90.
- Nunes LA, Grotto HZ, Brenzikofer R, Macedo DV (2014) Hematological and biochemical markers of iron status in a male, young, physically active population. Biomed Res Int 2014: 349182.
- Tsuboi A, Terazawa Watanabe M, Kazumi T, Fukuo K (2014) Serum copper, zinc and risk factors for cardiovascular disease in community-living Japanese elderly women. Asia Pac J Clin Nutr 23: 239-245.
- Liz Z, Trinidad D, Pittman EN, Riley EA, Sjodin A, et al. (2015) Urinary polycyclic aromatic hydrocarbon metabolites as biomarkers to woodsmoke exposure - results from a controlled exposure study. J Expo Sci Environ Epidemiol.
- Servaes K, Voorspoels S, Lievens J, Noten B, Allaerts K, et al. (2013) Direct analysis of phthalate ester biomarkers in urine without preconcentration: method validation and monitoring. J Chromatog A 1294: 25-32.
- Larsson K, Ljung Bj?rklund K, Palm B, Wennberg M, Kaj L, et al. (2014) Exposure determinants of phthalates, parabens, bisphenol A and triclosan in Swedish mothers and their children. Environ Int 73: 323-333.
- Kasapis C, Thompson PD (2005) The effects of physical activity on serum C-reactive protein and inflammatory markers: a systematic review. J Am Coll Cardiol 45: 1563-1569.
- Mannino DM, Tal-Singer R, Lomas DA, Vestbo J, Graham Barr R, et al. (2015) Plasma Fibrinogen as a Biomarker for Mortality and Hospitalized Exacerbations in People with COPD. Chronic Obstr Pulm Dis (Miami) 2: 23-34.
- Ridker PM, Hennekens CH, Buring JE, Rifai N (2000) C-reactive protein and other markers of inflammation in the prediction of cardiovascular disease in women. N Engl J Med 342: 836-843.
- Pagano G, Guida M, Tommasi F, Oral R (2015) Health effects and toxicity mechanisms of rare earth elements-Knowledge gaps and research prospects. Ecotoxicol Environ Saf 115: 40-48.
- Czajka M, Sawicki K, Sikorska K, Popek S, Kruszewski M, et al. (2015) Toxicity of titanium dioxide nanoparticles in central nervous system. Toxicol In Vitro 29: 1042-1052.
- Setyawati MI, Tay CY, Leong DT1, (2015) Mechanistic Investigation of the Biological Effects of SiO??TiO? and ZnO Nanoparticles on Intestinal Cells. Small 11: 3458-3468.
- Culver AL, Ockene IS, Balasubramanian R, Olendzki BC, Sepavich DM, et al. Statin use and risk of diabetes mellitus in postmenopausal women in the Women's Health Initiative. Arch Intern Med 2012; 172(2):144-52.
- Zeliger H (2012) Statin use and risk of diabetes. Arch Intern Med 172: 896.
- Devlin AM, Ngai YF, Ronsley R, Panagiotopoulos C (2012) Cardiometabolic risk and the MTHFR C677T variant in children treated with second-generation antipsychotics. Transl Psychiatry 2: e71.
- Shiu WY, Ma KC. (2000) Temperature dependence of physical-chemical properties of selected chemicals of environmental interest. I. Mononuclear and polynuclear aromatic hydrocarbons. J Phys Chem Ref Data 29: 41-66.
- Gordon CJ (2010) Response of the thermoregulatory system to toxic insults. Front Biosci (Elite Ed) 2: 293-311.
- Nakano Y (2007) Effect of chronic topical exposure to low-dose noxious chemicals and stress on skin sensitivity in mice. J Occup Health 49: 431-442.
- Gordon CJ, Johnstone AF, Aydin C (2014) Thermal stress and toxicity. Compr Physiol 4: 995-1016.
- Adachi M, Liu Y, Fujii K, Calderwood SK, Nakai A, et al. (2009) Oxidative stress impairs the heat stress response and delays unfolded protein recovery. PLoS One 4: e7719.
- Barrett MJ, Alones V, Wang KX, Phan L, Swerdlow RH (2004) Mitochondria-derived oxidative stress induces a heat shock protein response. J Neurosci Res 78: 420-429.
- Patterson RRM, Lima N. (2010) How will climate change affect mycotoxins in food. Food Res Int 43: 1902-14.