Keywords
Gene Expression; Pancreatic Neoplasms; Survival
Abbreviations
CSCs cancer stems cells; PDAC pancreatic ductal
adenocarcinoma; RNA Ribonucleic acid
INTRODUCTION
Pancreatic cancer is a challenging disease with dismal
prognosis for the vast majority of afflicted patients.
The incidence of pancreatic cancer in the United States
is estimated to be 48,960 in 2015 with 40,560 deaths
and a 5-year survival rate of 7% [1]. Pancreatic ductal
adenocarcinoma (PDAC) accounts for about 90% of the
malignant cancers arising from the ductal epithelium in
the exocrine part of the pancreas gland. Due to the silent
nature of the disease, most patients present late with
unresectable disease but approximately 20% will undergo
resection followed by chemotherapy and radiation
treatment. Disappointingly, despite the use of adjuvant
therapy, a significant number of these patients will recur
early after resection and die of the disease within one year [2] whereas 25% of these patients with resected PDAC can
live for 5 years or more [3]. Traditional prognostic factors
including stage, tumor grade, negative surgical margins,
and absence of lymph nodes cannot always accurately
predict long-term survival. The biology of the tumor may
be more important in predicting distant recurrence and
ultimately survival. Identifying prognostic factors that can
predict which patients may live longer would help with
treatment decisions postoperatively but also including the
use of neoadjuvant therapy as a means of delaying surgery
in patients who would otherwise have rapid disease
progression.
One way to select these individuals would be to
define a prognostic signature that can identify patients
with more aggressive tumor biology prior to treatment.
Many different aspects of PDAC tumor biology have been
suggested as candidates for determining the aggressive
phenotype including genetic [4], epigenetic [5], tumor
microenvironment [6], immune response [7] or presence
of cancer stem cells (CSCs) [8]. A recent report failed to
find a difference in the somatic mutation profile of PDACs
in very long-term survivors compared to PDACs in patients
unselected for survival [9]. In the absence of a specific
genetic mutation profile that discriminates long-term
survivors, another approach is to study gene expression. Expression profiling of PDAC has been undertaken by
several different investigators [10, 11] and uncovered
various signaling pathways associated with tumor
progression and metastatic disease. Of particular interest
is the study of Stratford et al. [12] who discovered a sixgene
signature that was predictive of survival in localized
PDAC in comparison to metastatic PDAC. Interestingly,
most genes in the classifier (SIGLEC11, KLF6, NFKBIZ,
ATP4A, GSG1, and FOSB) did not have an obvious role in
carcinogenesis, and only three had significantly higher
expression in the poor prognostic patients.
Instead of comparing primary PDAC tumors at the
extremes of disease (localized versus metastatic), we
specifically selected a subgroup of patients who were all
considered candidates for surgical resection, and from this
cohort we further selected patients with short-term (<10
months) and those with long-term survival >20 months).
Our focus was not only to identify genes of interest so
as to assist in the development of a specific prognostic
gene signature that could guide treatment decisions at
presentation and postoperatively but also to identify the
key pathways involved in patients with poor outcomes as
potential targets for novel treatment strategies.
Methods
Patient Consent and Sample Acquisition
Between February 2009 and November 2013, patients
who underwent a pancreaticoduodenectomy for pancreatic
adenocarcinoma were approached to submit portions of
their tumor to the Beaumont BioBank. A single surgeon
completed all resections. In addition to surgery, patients
received adjuvant therapy as previously reported [13].
Patients were consented by Beaumont BioBank clinical
staff using an IRB approved protocol (HIC 2008-180),
and samples were processed and stored at -80°C using
standard operating procedures. Inclusion criteria required
survival of greater than 100 days following surgery in
order to eliminate death due to surgical complications or
other comorbidities. Analysis was limited to patients who
did not present with distant metastasis and did not receive
preoperative chemo- or radiation therapy. Resection
margins were assessed using a standardized pathology
protocol based on axial specimen slicing and reporting
margin involvement if tumor cells are present within 1-2
mm from the margin. 39 patients were consented and their
specimens banked and of these 32 satisfied the inclusion
criteria. The overall median survival of all patients was
10.1 months. For this study, we were specifically interested
in whether there were genomic differences between the
longer-term survivors and those with shorter survival.
Therefore, we arbitrarily selected two sub-groups of
patients. We identified 11 patients who lived greater than
20 months following their surgery and 13 patients who
lived less than 10 months. This lower cut-off was chosen
based on the median survival and the upper cut-off of 20
months was simply a doubling of the median survival and
also gave a similar number of patients in each group.
RNA Isolation
Frozen pancreatic adenocarcinoma tissue specimens
stored at -80°C in RNAlater Stabilization Solution (Life
Technologies, Carlsbad, CA) were homogenized into lysis
buffer using the gentleMACS dissociator’s (Miltenyi Biotec
Inc., Auburn, CA) “Homogenization of tissue for total
RNA isolation” protocol. Following the manufacturer’s
protocol, RNA was purified using the E.Z.N.A. Total RNA
Kit I (Omega, Norcross, GA), quantified (Nanodrop 8000,
Thermo Scientific), and then stored at ‑80°C. RNA integrity
was determined by Bioanalyzer analysis (Agilent) just
prior to processing for expression microarray analysis.
Illumina Expression Beadchips
RNA was amplified and labeled using the TargetAmp-
Nano Labeling Kit (Epicenter, Madison, WI) which enables
amplification and target preparation compatible with
the Direct Hybridization Assay (Illumina, San Diego, CA).
Amplification was performed with 500 ng of total RNA
input following procedures described in the TargetAmp-
Nano Labeling Kit user guide. Hybridization and staining
to the HumanHT-12 v4 Expression BeadChip (Illumina,
San Diego, CA) was performed using 750 ng of BiotinaRNA
product following protocols outlined in the Whole-
Genome Gene Expression Direct Hybridization Assay Guide.
Subsequent scanning of the BeadChip was performed using
the iScan Microarray Scanner (Illumina, San Diego, CA).
Gene Expression and Pathway Analysis
Gene expression data from 24 samples were imported
into Illumina’s Genome Studio. They were imported with
cubic spline normalization. Quality control was performed
in Genome Studio. The Partek Report Plug-in from Illumina’s
Genome Studio was used to export the gene expression
data from 26 arrays into Partek’s Genomics Suite (version
6.15.1207). Differentially expressed genes were detected by
ANOVA (p≤0.01 and 2-fold cutoff) taking into account the
parameters of survival and barcode. Barcode refers to the chip
used; it is included to account for hybridization differences
associated with runs on different bead chips. Pathway
analysis was done with Pathway Studio (Elsevier, version
11.1.0.6 2015-12-08). The data are available using NCBI Gene
Expression Omnibus accession number GSE77435 [14].
Survival Analysis
Selected genes were analyzed for their influence
on survival from pancreaticoduodenectomy. The log 2
intensity values for the long and short-term survivors were
used to classify patients as above and below the median
value and analyzed using the Kaplan-Meier method and log
rank test. A p-value of <0.05 was considered statistically
significant. Statistical analyses were performed SPSS
(version 22; IBM SPSS, Amok, NY, USA)
RESULTS
Patient Clinical Data
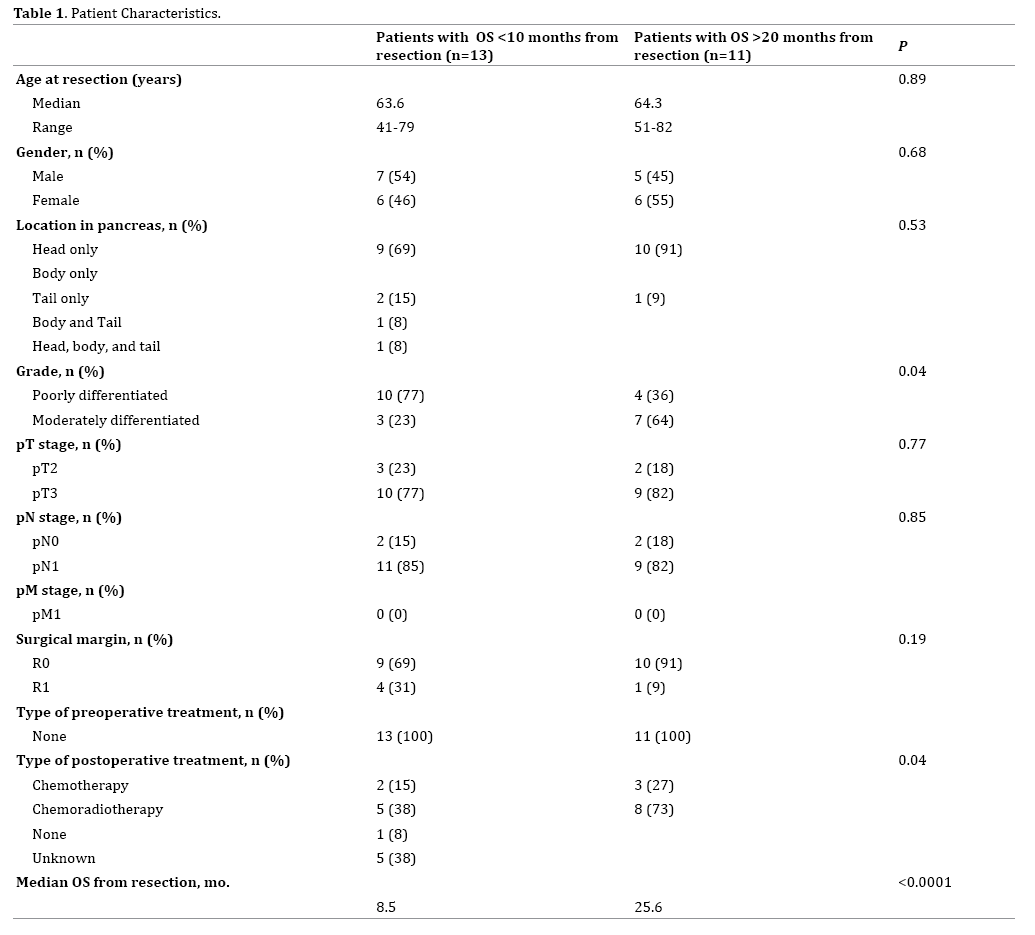
Table 1 lists patient characteristics, pathologic
findings, additional treatments, and overall survival (OS) from the time of surgery. The median age was 64 years,
and 50% of patients were male. The median OS after
surgery for all patients in the study was 10.1 months with
a median of 25.6 months and 8.5 months for the >20
months and <10 months groups, respectively. There was
no significant difference in the pathologic T (T3=77% vs.
82%; p=0.77) or N (N1=85% vs. 82%; p=0.85) stage or
positive surgical margins (31% vs. 9%; p=0.19) between
patients based upon OS. However, there was significantly
higher proportion of grade 3 tumors in the short-term
survivors compared to the long-term survivors (77% vs.
36%; p=0.04). There was also a difference in postoperative
(p=0.04) treatment.
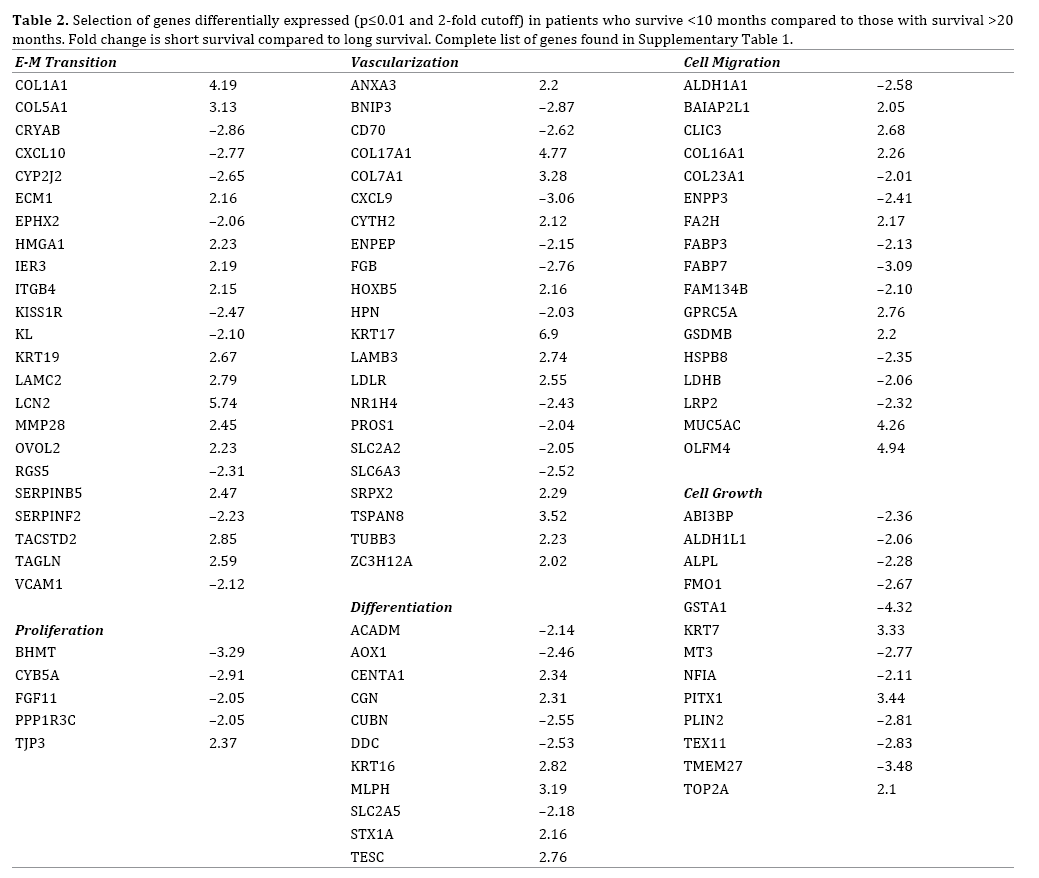
Gene Expression Differences At >20 Months
We identified 163 genes that were differentially
expressed (p≤0.01 and 2-fold cutoff) between patients
who survived <10 months and patients with survival
>20 months (Supplemental Table 1). This included genes associated with epithelial to mesenchymal transition,
vascularization, and cell migration (Table 2). Some of the
greatest increases in expression for short-term survivors
were seen in KRT17, S100P, LCN2, COL17A1, and COL1A1 amongst others whilst some of the most prominent genes
that were downregulated in the long-term survivors
included GSTA1, GSTA2, LGALS2, and CXCL9.
Signaling Changes At >20 Months
Pathway Studio utilizes a literature mining tool,
MedScan, to generate sub-networks that associate genes
with other entities such as cell processes. A Fisher’s Exact
test was used to identify sub-networks that are highly
represented by differentially expressed genes. One type
of sub-network associates genes with a central seed based
upon the ability of the seed to control gene expression.
The top expression target sub-networks that were highly
represented by the differentially expressed genes included
the expression targets of SP1, JUN, and PPARG (Figure 1).
Figure 1. Combined expression target sub-networks of SP1, JUN, and PPARG identified as highly represented in the list of genes differentially expressed (p≤0.01 and 2-fold) between patients who survived <10 months and those that survived >20 months. Genes in red are upregulated in patients with shorter survival; genes in blue, downregulated.
Of note is the general downregulation of PPARG expression
targets and the presence in these sub-networks of COL1A1,
COL7A1, GPRC5A, KRT17, and ECM1 which have not been
previously linked to patient outcomes in PDAC. Another type
of sub-network includes genes involved in regulating cell
processes. Differentially expressed genes between patients
with OS <10 months and those with OS >20 months are
highly represented by genes regulating cell differentiation,
cell proliferation, cell migration, and vascularization.
Survival Analysis
Figure 2a shows the Kaplan-Meier survival curves for
ALDH1A1, COL17A1, COL7A1 and KRT17 selected from Table 1 and 2. COL17A1 and COL7A1 were the most
significant genes in terms of overexpression being linked
to poor survival (Figure 2b, 2c) whilst ALDH1A1 was the
most significant gene where underexpression was linked
to shorter survival (Figure 2a).
Figure 2. Kaplan-Meier survival analyses of selected genes. The green solid lines represent tumors with lower than median expression whilst the red dotted lines are those with higher than median expression. The genes shown are (a). ALDH1A1, (b). COL17A1, (c). COL7A1 and (d). KRT17. The p-values represent the Log-Rank test.
DISCUSSION
This study reports on pancreatic adenocarcinoma gene
expression differences in patients who survived greater than 20 months or less than 10 months following surgery. There
was no significant difference in the age, sex, or stage between
the two groups, and no one received preoperative therapy.
There was, however, a significantly greater percentage of
high grade tumors in patients who lived <10 months.
Traditional prognostic criteria for long-term survivors
have included negative margin status, small tumor size,
no lymph node involvement, low CA 19-9 level, low grade,
absence of metastases, and type of treatment administered
[15, 16, 17]. However, the use of these prognostic factors
has limited value due to the heterogeneity of the long-term
survival group. Ferrone et al. showed that negative margins
and negative nodes demonstrated a positive prognosis, but
at the same time 41% of long-term survivors had positive
nodes and 24% had positive margins [18]. Adham et al. found
that typically positive prognostic criteria did not predict long
survival with 29 of 30 long-term survivors having T3/T4
tumors with 12 of 30 having positive lymph nodes [19].
One possible conclusion is that the biology of the
tumor, not traditional prognostic markers, is important for
prediction of long-term survival. One recent study by Dal Malin et al. tried to address this by using next-generation
exome sequencing to examine the genomic profile of longterm
survivors [9]. While mutations were found in KRAS,
TP53, SMAD4, and CDKN2A, there were no mutations that
were preferentially found in the long-term survivors.
In order to continue the search for the biological
variability seen in the long-term survivors, we have
identified 163 genes that were differentially expressed
between the <10 months and >20 months survival
groups. Several of the genes we identified have a known
prognostic role in pancreatic adenocarcinoma including
ADAM metallopeptidase domain 8 (ADAM8) and transgelin
(TAGLN) along with aldehyde dehydrongenase 1 family,
member A1 (ALDH1A1). However, most of the genes we
identified have not been previously linked to prognosis in
pancreatic adenocarcinoma.
Aldehyde dehydrogenase 1 family, member A1 is an
enzyme involved in alcohol metabolism. It was found to have
decreased expression (2.6-fold) in short-term survivors
and was significant in overall survival analysis. This gene
has been previously linked to prognosis and progression
in many cancers [20, 21, 22, 23]. Expression of this gene
has also been linked to cancer stem cells (CSCs), with low
expression associated with gemcitabine resistance and
poor prognosis in pancreatic adenocarcinoma [24, 25, 26].
Keratin 17 is an intermediate type I filament chain
keratin usually expressed in the nail bed, hair follicle, and
sebaceous glands. This gene was found to have higher
expression (6.9 fold) in in the short-term OS patients
and was significant in overall survival analysis. Increased
expression of this gene has been linked to poor survival
in cervical squamous cell carcinoma, epithelial ovarian
cancer, gastric cancer, and breast cancer [27, 28, 29, 30].
However, this has not been previously demonstrated to be
prognostic in pancreatic cancer.
Several members of the COL family were associated
with short survival. The COL (collagen) family comprises
28 members that contain at least one triple-helical domain.
Collagens are deposited in the extracellular matrix (ECM)
where most of them form supramolecular assemblies.
They are known to affects the tumor microenvironment
through degradation and re-deposition contributing
to ECM remodeling which promotes tumor infiltration,
angiogenesis, invasion and migration. While collagen
was traditionally regarded as a passive barrier to
resist tumor cells, it is now evident that collagen is also
actively involved in promoting tumor progression [31].
COL17A1overexpression was the most significant gene
in terms of overexpression being associated with shorter
survival. This gene has also been recently identified as
a potential biomarker using a minimum-redundancymaximum-
relevance (mRMR) method interrogating a set
of transcriptome data of pancreatic cancer [32].
This study adds to the growing literature on the
prognostic utility of gene expression patterns for PDAC.
The University of Virginia recently published a 13-gene signature that predicts significantly higher risks of
mortality in pancreatic adenocarcinoma patients [33].
Of the 13 genes these authors identified, 4 including TGFA, ELAVL1, and MDM2 had been previously shown
to be important in pancreatic adenocarcinoma, 6 genes
were associated with prognosis or highly expressed in
other forms of cancer but not previously reported in
PDAC, and 3 had not been reported to be prognostically
significant in any malignancy. Those categorized as low
risk by this gene signature had a median overall survival
of 14 months compared to 6 months for high risk patients.
There was no overlap in the genes identified for their gene
signature and the genes identified in the current study.
A 6-gene prognostic signature was also published by the
University of North Carolina [12]. This signature included
FOSB, NFKBIZ, IKBZ, MAIL, GSGI, and SIGLEC11. Patients
classified as low risk by this study had a median overall
survival of 49 months, and those classified as high risk had
a median overall survival of 15 months.
Additionally the current results were compared to a
pair of studies in NCBI’s Gene Expression Omnibus. The
original purpose of these published studies was not to
examine survival specifically, but both studies included
survival data for at least some of the publicly available
data. In a study by Van den Broeck et al. [34], microarrays
(GEO accession GSE42952) were used to compare patients
with good (DFS>50 months) and poor (DFS<7 months)
outcome. From a study by Yang et al. [35], the data from
a subset of patients (GEO accession GSE62452) were used
to compare patients with similar outcomes as the current
study – patients with OS>20 months or <10 months. Of the
genes that were identified as differentially expressed
in the current study (Supplemental Table 1), 56 were
validated through the analysis of the data in these
studies. This included genes such as COL1A1, COL5A1,
COL7A1, CYB5A, and STX1A that were confirmed by
both studies. Even more striking was the concordance
in regulated cell processes between our current study
and the publicly available data. Of the highly regulated
cell processes that we identified, five of the top six
were found in both external studies. This included
cell differentiation, cell invasion, cell proliferation,
cell migration, and cell behavior which were all highly
ranked in both publicly available datasets.
In addition to individual gene expression biomarkers,
signaling surrounding SP1, JUN, and EGF was highly
altered in the patients that showed longer survival. This
result confirmed the role of these signaling pathways
in pancreatic cancer. SP1 is a negative prognostic factor
that plays a role in cell proliferation and metastasis [36].
In particular, SP1 protein was found to be overexpressed
in a subset of primary pancreatic adenocarcinoma that
developed lymph node metastasis, were higher stage and
grade, and had a much shorter overall survival [37]. JUN is
a transcription factor involved with cell proliferation that
has been identified as an oncogene. It has previously been
related to pancreatic cancer stage, grading, and invasion
[38]. Expression of JUN was shown to be elevated in liver metastases compared to pancreatic cancer tissue, and high
expression was seen more often in short-term survivors
[39]. EGF acts via its receptor (EGFR) to potentiate growth,
proliferation and differentiation of many different cell
types. Specifically, it has been shown to be involved in
growth, invasiveness, and metastasis of pancreatic cancer
[40, 41].
In this comparison of patients who live <10 months and
>20 months following definitive resection for pancreatic
adenocarcinoma, we have identified multiple differentially
expressed genes. Some of these genes have previously been
shown to be prognostic in pancreatic adenocarcinoma, but
most had never been linked to survival in these patients.
These genes and their expression targets warrant further
investigation to determine their value as prognostic
markers or targets for molecular therapy.
Acknowledgements
This work was funded by the Mopper Family
philanthropy fund.
Conflicts of interest
The authors declare that they have no conflicts of
interest.
References
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin 2015;65:5-29. [PMID: 25559415].
- Neoptolemos JP, Stocken DD, Friess H, Bassi C, Dunn JA, Hickey H, et al. A randomized trial of chemoradiotherapy and chemotherapy after resection of pancreatic cancer. N Engl J Med 2004;350:1200-1210. [PMID: 15028824].
- Ferrone CR, Brennan MF, Gonen M, Coit DG, Fong Y, Chung S, et al. Pancreatic adenocarcinoma: the actual 5-year survivors. J Gastrointest Surg 2008;12:701-706. [PMID: 18027062].
- Wood LD, Hruban RH. Genomic landscapes of pancreatic neoplasia. J Pathol Transl Med 2015;49:13-22. [PMID: 25812653].
- Neureiter D, Jager T, Ocker M, Kiesslich T. Epigenetics and pancreatic cancer: pathophysiology and novel treatment aspects. World J Gastroenterol 2014;20:7830-7848. [PMID: 24976721].
- Xu Z, Pothula SP, Wilson JS, Apte MV. Pancreatic cancer and its stroma: a conspiracy theory. World J Gastroenterol 2014;20:11216-11229. [PMID: 25170206].
- Inman KS, Francis AA, Murray NR. Complex role for the immune system in initiation and progression of pancreatic cancer. World J Gastroenterol 2014;20:11160-11181. [PMID: 25170202].
- Tanase CP, Neagu AI, Necula LG, Mambet C, Enciu AM, Calenic B, et al. Cancer stem cells: involvement in pancreatic cancer pathogenesis and perspectives on cancer therapeutics. World J Gastroenterol 2014;20:10790-10801. [PMID: 25152582].
- Dal Molin M, Zhang M, de Wilde RF, Ottenhof NA, Rezaee N, Wolfgang CL, et al. Very Long-term Survival Following Resection for Pancreatic Cancer Is Not Explained by Commonly Mutated Genes: Results of Whole-Exome Sequencing Analysis. Clin Cancer Res 2015;21:1944-1950. [PMID: 25623214].
- Grutzmann R, Boriss H, Ammerpohl O, Luttges J, Kalthoff H, Schackert HK, et al. Meta-analysis of microarray data on pancreatic cancer defines a set of commonly dysregulated genes. Oncogene 2005;24:5079-5088. [PMID: 15897887].
- Grutzmann R, Saeger HD, Luttges J, Schackert HK, Kalthoff H, Kloppel G, et al. Microarray-based gene expression profiling in pancreatic ductal carcinoma: status quo and perspectives. Int J Colorectal Dis 2004;19:401-413. [PMID: 14745573].
- Stratford JK, Bentrem DJ, Anderson JM, Fan C, Volmar KA, Marron JS, et al. A six-gene signature predicts survival of patients with localized pancreatic ductal adenocarcinoma. PLoS Medicine 2010;7:e1000307. [PMID: 20644708].
- Baschnagel A, Shah C, Margolis J, Nadeau L, Stein J, Jury R, et al. Survival after chemoradiation in resected pancreatic cancer: the impact of adjuvant gemcitabine. Int J Radiat Oncol Bio Phys 2012;83:e331-335. [PMID: 22420967].
- Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res 2002;30:207-210. [PMID: 11752295].
- Shimada K, Sakamoto Y, Nara S, Esaki M, Kosuge T, Hiraoka N. Analysis of 5-year survivors after a macroscopic curative pancreatectomy for invasive ductal adenocarcinoma. World J Surg 2010;34:1908-1915. [PMID: 20376443].
- Dusch N, Weiss C, Strobel P, Kienle P, Post S, Niedergethmann M. Factors predicting long-term survival following pancreatic resection for ductal adenocarcinoma of the pancreas: 40 years of experience. J Gastrointest Surg 2014;18:674-681. [PMID: 24241965].
- Paniccia A, Hosokawa P, Henderson W, Schulick RD, Edil BH, McCarter MD, et al. Characteristics of 10-Year Survivors of Pancreatic Ductal Adenocarcinoma. JAMA Surgery 2015; 150:701-710. [PMID: 26062046].
- Ferrone CR, Pieretti-Vanmarcke R, Bloom JP, Zheng H, Szymonifka J, Wargo JA, et al. Pancreatic ductal adenocarcinoma: long-term survival does not equal cure. Surg 2012;152:S43-49. [PMID: 22763261].
- Adham M, Jaeck D, Le Borgne J, Oussoultzouglou E, Chenard-Neu MP, Mosnier JF, et al. Long-term survival (5-20 years) after pancreatectomy for pancreatic ductal adenocarcinoma: a series of 30 patients collected from 3 institutions. Pancreas 2008;37:352-357. [PMID: 18665012].
- Goossens-Beumer IJ, Zeestraten ECM, Benard A, Christen T, Reimers MS, Keijzer R, et al. Clinical prognostic value of combined analysis of Aldh1, Survivin, and EpCAM expression in colorectal cancer. Br J Cancer 2014;110:2935-2944. [PMID: 24786601].
- Zenke Y, Ishii G, Ohe Y, Kaseda K, Yoshida T, Matsumoto S, et al. Aldehyde dehydrogenase 1 expression in cancer cells could have prognostic value for patients with non-small cell lung cancer who are treated with neoadjuvant therapy: identification of prognostic microenvironmental factors after chemoradiation. Pathol Int 2013;63:599-606. [PMID: 24422956].
- Ajani JA, Wang X, Song S, Suzuki A, Taketa T, Sudo K, et al. ALDH-1 expression levels predict response or resistance to preoperative chemoradiation in resectableesophageal cancer patients. Mol Oncol 2014;8:142-149. [PMID: 24210755].
- Wu Q, Shi H, Holm R, Li X, Trope C, Nesland JM, et al. Aldehyde dehydrogenase-1 predicts favorable prognosis in patients with vulvar squamous cell carcinoma. Anticancer Res 2014;34:859-865. [PMID: 24511023].
- Kahlert C, Bergmann F, Beck J, Welsch T, Mogler C, Herpel E, et al. Low expression of aldehyde dehydrogenase 1A1 (ALDH1A1) is a prognostic marker for poor survival in pancreatic cancer. BMC cancer 2011;11:275. [PMID: 21708005].
- Duong HQ, Hwang JS, Kim HJ, Kang HJ, Seong YS, Bae I. Aldehyde dehydrogenase 1A1 confers intrinsic and acquired resistance to gemcitabine in human pancreatic adenocarcinoma MIA PaCa-2 cells. Int J Oncol 2012;41:855-861. [PMID: 22710732].
- Kim MP, Fleming JB, Wang H, Abbruzzese JL, Choi W, Kopetz S, et al. ALDH Activity Selectively Defines an Enhanced Tumor-Initiating Cell Population Relative to CD133 Expression in Human Pancreatic Adenocarcinoma. PLoS One 2011;6:e20636. [PMID: 21695188].
- Escobar-Hoyos LF, Yang J, Zhu J, Cavallo JA, Zhai H, Burke S, et al. Keratin 17 in premalignant and malignant squamous lesions of the cervix: proteomic discovery and immunohistochemical validation as a diagnostic and prognostic biomarker. Modern Pathology 2014;27:621-630. [PMID: 24051697].
- Wang YF, Lang HY, Yuan J, Wang J, Wang R, Zhang X, et al. Overexpression of keratin 17 is associated with poor prognosis in epithelial ovarian cancer. Tumour Biol 2013;34:1685-1689. [PMID: 23430585].
- van de Rijn M, Perou CM, Tibshirani R, Haas P, Kallioniemi O, Kononen J, et al. Expression of cytokeratins 17 and 5 identifies a group of breast carcinomas with poor clinical outcome. Am J Pathol 2002;161:1991-1996. [PMID: 12466114].
- Ide M, Kato T, Ogata K, Mochiki E, Kuwano H, Oyama T. Keratin 17 expression correlates with tumor progression and poor prognosis in gastric adenocarcinoma. Ann Surg Oncol 2012;19:3506-3514. [PMID: 22695933].
- Fang M, Yuan J, Peng C, Li Y. Collagen as a double-edged sword in tumor progression. Tumour Biol 2014;35:2871-2882. [PMID: 24338768].
- Shen S, Gui T, Ma C. Identification of molecular biomarkers for pancreatic cancer with mRMR shortest path method. Oncotarget 2017;8:41432-41439. [PMID: 28611293].
- Newhook TE, Blais EM, Lindberg JM, Adair SJ, Xin W, Lee JK, et al. A thirteen-gene expression signature predicts survival of patients with pancreatic cancer and identifies new genes of interest. PLoS One 2014;9:e105631. [PMID: 25180633].
- Van den Broeck A, Vankelecom H, Van Eijsden R, Govaere O, Topal B. Molecular markers associated with outcome and metastasis in human pancreatic cancer. Journal of Experimental & Clinical Cancer Research 2012;31:68. [PMID: 22925330].
- Yang S, He P, Wang J, Schetter A, Tang W, Funamizu N, et al. A Novel MIF Signaling Pathway Drives the Malignant Character of Pancreatic Cancer by Targeting NR3C2. Cancer Res 2016;76:3838-3850. [PMID: 27197190].
- Vizcaino C, Mansilla S, Portugal J. Sp1 transcription factor: A long-standing target in cancer chemotherapy. Pharmacol Ther 2015;152:111-124. [PMID: 25960131].
- Jiang NY, Woda BA, Banner BF, Whalen GF, Dresser KA, Lu D. Sp1, a new biomarker that identifies a subset of aggressive pancreatic ductal adenocarcinoma. Cancer Epidemiol Biomarkers Prev 2008;17:1648-1652. [PMID: 18628415].
- Tessari G, Ferrara C, Poletti A, Dubrovich A, Corsini A, Del Favero G, et al. The expression of proto-oncogene c-jun in human pancreatic cancer. Anticancer Res 1999;19:863-867. [PMID: 10216507].
- Ferrara C, Tessari G, Poletti A, Giacon C, Meggiato T, Martines D, et al. Ki-67 and c-jun expression in pancreatic cancer: a prognostic marker? Oncology Reports 1999;6:1117-1122. [PMID: 10425312].
- Kolb A, Kleeff J, Arnold N, Giese NA, Giese T, Korc M, et al. Expression and differential signaling of heregulins in pancreatic cancer cells. Int J Cancer 2007;120:514-523. [PMID: 17096356].
- Pryczynicz A, Guzinska-Ustymowicz K, Czyzewska J, Kemona A. Expression of epidermal growth factors and apoptosis markers in pancreatic ductal adenocarcinoma. Folia Histochem Cytobiol 2009;47:667-671. [PMID: 20430737].



