Keywords
Cholangiopancreatography, Endoscopic Retrograde; Jaundice, Obstructive; Pancreatitis; Serology
Abbreviations
ERCP: endoscopic retrograde cholangiopancreatography; FNA: fine needle aspiration
INTRODUCTION
Several reports have described patients with chronic pancreatitis which is associated with autoimmune diseases. Chronic pancreatitis caused by an autoimmune mechanism has been variably termed as primary sclerosing pancreatitis [1], lymphoplasmacytic sclerosing pancreatitis or autoimmune pancreatitis [2]. Called autoimmune pancreatitis in 1995 by Yoshida et al. [2], the clinical characteristics of the disease had been described as early as 1961 [3]. Only recently has autoimmune pancreatitis been recognized as a distinct clinical entity. It is a multisystem disorder, often with extra-pancreatic manifestations, including immunoglobulin G4-associated cholangitis. It is one of the few autoimmune conditions which predominantly affects male subjects in the fifth and sixth decades of life. Obstructive jaundice is the most common presenting symptom with pancreatic carcinoma being the most important differential diagnosis. It can also present as acute or chronic pancreatitis [4]. In recent years, diagnostic criteria
autoimmune pancreatitis have been established, most of which rely on a combination of clinical presentation, imaging of the pancreas and other organs (by CT scan, MRI and endoscopic retrograde pancreatography), IgG4 serology, pancreatic histology and response to steroids [5, 6]. Recently, a novel antibody against the plasminogen-binding protein of Helicobacter pylori has been found in most patients with autoimmune pancreatitis [7]. The first diagnostic criteria were proposed by the Japan Pancreas Society in 2002 which were later modified in 2006 [8, 9]. These diagnostic criteria are important for differentiating autoimmune pancreatitis from its mimickers, such as pancreatic adenocarcinoma and primary sclerosing cholangitis. Recently, HISORt criteria have been proposed by the Mayo Clinic and include pancreatic histology (H), typical imaging (I), serology (S), other organ involvement (O) and response to steroid therapy (Rt) [5]. Accordingly, patients can be grouped into 3 groups: Group A includes diagnostic pancreatic histology, Group B includes typical imaging and positive serology and Group C includes patients with unexplained pancreatic disease with positive serology and/or other organ involvement with resolution/marked improvement in pancreatic/extrapancreatic manifestations with steroid therapy. Recently, Asian diagnostic criteria have also been defined in a Japan-Korea symposium on autoimmune pancreatitis [10].
After the first description from Japan, autoimmune pancreatitis has been reported from a number of countries but the exact incidence is not known. There are only a few reports from South Asia. With the present report we wish to point out that lack of awareness of the disease may be responsible for this lack. It is interesting to know that three countries (Japan, Korea and Italy) have shown comparable prevalence rates (4.6-6% of chronic pancreatitis) in spite of different environmental and genetic backgrounds [11].
METHODS
Patients
A retrospective analysis of all cases of autoimmune pancreatitis seen by us in the last three years (July 2006 - June 2009) was carried out. Records of all patients with a diagnosis of autoimmune pancreatitis were retrieved and analyzed. Diagnosis of autoimmune pancreatitis was established on the basis of imaging, serology, supportive cytology and response to treatment. Details about clinical presentation were retrieved, and the time gap between presentation and final diagnosis was recorded. All patients had undergone a work-up for pancreatitis which included abdominal contrast-enhanced computed tomography (CECT) and endoscopic retrograde cholangiopancreatography (ERCP). As the diagnosis of autoimmune pancreatitis was not suspected clinically in all the patients in the first evaluation, they were treated as per the clinical indication until the correct diagnosis of autoimmune pancreatitis was established. For the same reason, serology was carried out after a variable gap in clinical presentation in the first three patients in which follow-up radiology initially suggested autoimmune pancreatitis, and subsequent serology confirmed the diagnosis.
Imaging Techniques
Contrast-enhanced computed tomography (CECT) was carried out using a 16-slice multidetector computed tomographic scanner (Somatom sensation®, Siemens, Siemens, Forchheim, Germany). Data acquisition was done in the portovenous phase (65-70 sec of delay) from domes of diaphragm till pubic symphysis with negative oral contrast (2 L mineral water) and 80-100 mL of intravenous non-ionic water-soluble iodinated contrast media. Data were analyzed on retroreconstructed thin (2 mm) axial sections and in 3D reformatted planes.
Endoscopic retrograde cholangiopancreatography (ERCP) examinations were carried out using a side viewing endoscope (Olympus Corporation, Tokyo, Japan). Water-soluble iodinated contrast was used for opacification of the pancreatic and bile ducts. Three to six spot films were taken for each patient.
Image Analysis
CECT images were analyzed for any of the following features:
a. pancreas enlargement (diffuse or focal) with or without a hypoattenuating rim, dilated pancreatic duct, pancreatic atrophy, pancreatic calcification;
b. biliary system (extra/intra hepatic) or hilar biliary strictures;
c. other organ involvement (kidneys), lymphadenopathy (mediastinal/hilar/abdominal), retroperitoneal fibrosis or any other abnormal finding.
Cholangiopancreatogram images were analyzed for any of the following features:
a. biliary strictures (site) and extent of narrowing;
b. pancreatic duct (stricture) and extent of dilatation.
Serum Immunoglobulin G4
Serum immunoglobulin G4 was estimated using the BINDARIDTM (The Binding Site Limited, Birmingham, United Kingdom) human IgG subclass estimation from the binding site by a single radial immunodiffusion method using the method described by Mancini et al. [12]. Five μL of the serum sample were diluted 1/5 times with 7% bovine serum albumin, loaded into the wells and incubated for 72 hours at room temperature for the complete development of the precipitin rings. Serum IgG4 values were calculated by measuring the diameter of the rings and reading the values from a table provided with the kit for this purpose. Serum IgG4 level greater than 119 mg/dL were considered to be suggestive of autoimmune pancreatitis [13].
Cytology
Fine needle aspiration (FNA) was used to diagnose tissue from the pancreas under ultrasound guidance by using the percutaneous transabdominal route in 4 patients and endoscopic brushings from the papilla were taken in one patient. The smears were processed after air drying and fixation in alcohol. They were stained with May-Grunwald Giemsa stain and Hematoxylin and Eosin stain, respectively. Cytologically, the presence of lymphocytes, plasma cells and stromal fibroinflammatory fragments with embedded lymphocytes admixed with variable numbers of pancreatic ductal and acinar cells strongly supported a diagnosis of autoimmune pancreatitis. Furthermore, the absence of malignant cells was documented.
Response to Steroid Therapy
Prednisolone was started at a dose of 40 mg/day for 4 weeks followed by a tapering off of 5 mg per week over the next 7 weeks. Periodic follow-ups were carried out in all patients at two-week intervals. Follow-up visits included clinical evaluation, liver function testing and imaging.
ETHICS
The principles of the Declaration of Helsinki were adhered to throughout this study.
STATISTICS
Absolute frequency was used as a descriptive statistic.
RESULTS
Out of 195 patients with chronic pancreatitis admitted to our department between July 2006 and June 2009, five patients were diagnosed as having autoimmune pancreatitis (2.6%) in this 3-year period. Clinical presentation and proof of diagnosis for all 5 patients are reported in Tables 1 and 2. Four of the 5 patients were males. The most common presenting symptom was cholestatic jaundice (5/5) followed by abdominal pain (3/5) and weight loss of 3-5 kg (3/5). The most common initial diagnosis was pancreatic head mass (3/5). The first two patients (Cases #1 and #2), with an initial diagnosis of pancreatic head mass, underwent biliary stenting. It was only on follow-up imaging that the radiologist suspected autoimmune pancreatitis and subsequent serology and cytology were evaluated to confirm the diagnosis of autoimmune pancreatitis. One of the patients (Case #3) had been diagnosed as primary sclerosing cholangitis 7 years earlier on the basis of cholangiographic features showing a dominant stricture in the common bile duct for which he underwent biliary stenting several times. During the follow-up, the diagnosis of autoimmune pancreatitis was suspected and finally, the diagnosis of autoimmune pancreatitis was established on the basis of endoscopic brush cytology from the papilla and IgG4 serology.

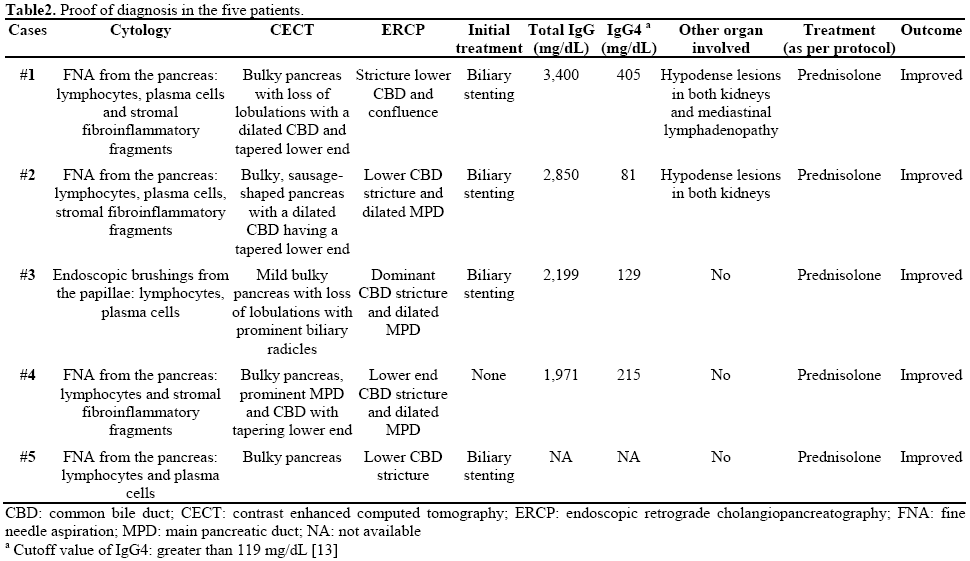
One patient (Case #4) had had obstructive jaundice and abdominal pain for 3 months. CECT abdomen suggested the diagnosis of autoimmune pancreatitis which was confirmed by serology and cytology. The last patient (Case #5) had had obstructive jaundice (serum bilirubin greater than 10 mg/dL; reference range: 0.3-1.0 mg/dL) and abdominal pain for 3 months. His abdominal ultrasound had shown features of pancreatic head mass. He underwent biliary stenting. However, an abdominal CECT suggested autoimmune pancreatitis. He was started on steroids to which he responded dramatically. IgG4 serology could not be obtained in his case.
Tissue diagnosis was achieved in all 5 patients on the basis of percutaneous FNA cytology (Cases #1, #2, #4, and #5) and papillary brush cytology (Case #3). The cytological findings included the presence of pancreatic ductal and acinar cells, lymphocytes, plasma cells and stromal fibroinflammatory fragments with lymphocytes (Figure 1). Immunostaining for IgG4 was also performed on the aspiration smears but was not contributory, probably due to technical reasons or the nature of the specimen.
Figure 1: Panel showing fine needle aspiration cytological features in autoimmune pancreatitis. a. Pancreatic ductal epithelial fragments and stromal
fibroinflammatory fragments at low magnification. b. Pancreatic acinar cell cluster with lymphocytes and plasma cells in the background. c. Cellular
stromal fibroinflammatory fragment. d. Hyalinised stromal fragment. (a., c., d.: Hematoxyline-Eosin stain; b.: May-Grünwald Giemsa stain; a.:
x100; b.: x200; c. and d.: x400).
The most common findings on imaging were bulky pancreas with loss of lobulations and common bile duct stricture, and the majority (4/5) underwent common bile duct stenting (Figures 2 and 3). IgG4 serology was carried out in 4 of the 5 patients and it was positive in 3 of them. Two of the patients had extrapancreatic manifestations in the form of hypodense lesions in the kidney (2/5) and mediastinal lymphadenopathy (1/5). All patients were treated with prednisolone 40 mg/day for 4 weeks followed by a tapering off of 5 mg per week over the next 7 weeks; all of them showed rapid clinical improvement. Follow-up liver function tests showed resolution of hyperbilirubinemia and a decrease in serum alkaline phosphatase. Patients with common bile duct stricture underwent repeat ERCP and their cholangiograms showed resolution of the stricture following therapy with steroids (Figure 3).
Figure 2: Contrast-enhanced computed tomographic axial image
shows a diffusely enlarged pancreas with loss of lobulations. There
are multiple hypoattenuating lesions seen in the left kidney (thin
arrows). Note dilated intrahepatic biliary radicals (thick arrows) and
plastic common bile duct stent in-situ (curved arrow).
Figure 3: a. ERCP image prior to treatment shows a long segmental
smooth stricture at the terminal end (long arrow) with a dilated
common bile duct. There is another tight stricture at confluence
involving both right and left biliary ducts (small arrow) with dilated
biliary radicals in both lobes of the liver. b. Post-treatment ERCP
image shows resolution of the strictures with a common bile duct of
normal caliber and non-dilated biliary radicals.
There was no recurrence after a follow-up of 6-8 months.
DISCUSSION
Autoimmune pancreatitis is a relatively newly characterized disease entity with most of the knowledge only having been gained in the last decade. It was first reported in Japan but now cases have been reported in Europe, United States, China and Australia [5, 14, 15, 16, 17]. Reports from Africa and the Middle East are, however, lacking. Three cases have been reported from two centers in India in which the diagnosis was based upon imaging and response to steroids [11, 18]. In the present report, the diagnosis was based on standard criteria with serology, radiology and cytopathological findings, and a dramatic response to corticosteroid therapy. All 5 of our patients fulfilled both the HISORt [5] and the Asian diagnostic criteria [10].
Autoimmune pancreatitis is most commonly misdiagnosed as carcinoma in the head of the pancreas because the presenting features of these two diseases are similar. It is imperative to differentiate autoimmune pancreatitis from pancreatic cancer owing to the vastly different prognostic and therapeutic implications. Even in Japan, all the cases were initially diagnosed as pancreatic cancer in the earlier reported series [19]. In a series of autoimmune pancreatitis from the United Kingdom, the majority of patients (73%) were referred with suspected pancreatic malignancy on cross sectional imaging [20]. In a Korean study of 67 patients, 12 patients underwent surgery due to a diagnosis of pancreatic carcinoma [21]. The focal mass-forming type of pancreatitis is difficult to differentiate from pancreatic cancer. It is in this situation that obtaining a tissue diagnosis becomes imperative. This may be done by either core needle biopsy or FNA cytology through the percutaneous transabdominal approach or by endoscopic ultrasound. The cytopathological features in autoimmune pancreatitis are well described in a series presented by Deshpande et al. in 2005 [22] and which were corroborated by us in this series of cases. The findings of stromal fibroinflammatory fragments with embedded lymphocytes and a lymphoplasmacytic infiltrate support a diagnosis of autoimmune pancreatitis in the right clinical context and with the correct imaging findings [22].
Imaging findings suggestive of autoimmune pancreatitis are the presence of focal or diffuse enlargement of the gland (sausage-shaped) with loss of lobulations with or without a capsule-like rim and the absence of vascular encasement or calcification [23, 24, 25]. Extrapancreatic manifestations are quite common in autoimmune pancreatitis and, if present, can aid in strengthening the diagnosis. The biliary tract is the most common extrapancreatic system to be involved in autoimmune pancreatitis (30-90%). Both intrahepatic and extrahepatic bile ducts can be affected; however, the distal common bile duct is the most common site of involvement [23, 24, 25]. Renal involvement is also common (35%) in the form of multiple small hypoattenuating peripheral cortical rounded wedge-shaped nodules [26]. Other extrapancreatic manifestations in autoimmune pancreatitis include hilar lymphadenopathy, lacrimal and salivary gland involvement and retroperitoneal fibrosis [24, 25]. The hallmark finding on ERCP in patients with autoimmune pancreatitis is a focal, diffuse or segmental attenuation of the main pancreatic duct and the disappearance of right-angled branches. The main pancreatic duct adjacent to or upstream of the strictures is minimally dilated. Other common findings on ERCP are the narrowing of the intrapancreatic portion of the common bile duct, irregular narrowing of the extrahepatic bile ducts and, less frequently, enlarged intrahepatic bile ducts [25, 27]. Pancreatographic findings were available in 3 of our patients and they all showed uneven contour and dilatation of the main pancreatic duct.
One of our patients had been diagnosed as a case of primary sclerosing cholangitis 7 years earlier on the basis of a cholangiogram showing a dominant biliary stricture. The diagnosis of autoimmune pancreatitis was finally established on the basis of serology and papillary brush cytology. Primary sclerosing cholangitis is a common mimicker of autoimmune pancreatitis/cholangitis. A report of 192 patients with primary sclerosing cholangitis from Japan showed a high incidence of chronic pancreatitis and positive antinuclear antibodies along with a lower incidence of ulcerative colitis in elderly men [28]. Some of these patients would fit in with the diagnosis of autoimmune pancreatitis. Differentiating primary sclerosing cholangitis from autoimmune cholangitis is challenging. In a recent study, swollen duodenal papillae and IgG4-positive plasma cells in the duodenal papillae were more common in patients of autoimmune pancreatitis than in patients with primary sclerosing cholangitis [29]. Another study noted segmental stenosis of the lower common bile duct only in autoimmune pancreatitis while a beaded or “prunedtree” appearance was detected only in primary sclerosing cholangitis patients and the response to steroids was better in autoimmune pancreatitis [30].
their diagnosis and the scarcity of reports from Africa and South East Asia suggest that lack of awareness about autoimmune pancreatitis may be responsible for the underreporting of the disease. While the prevalence of autoimmune pancreatitis is 6% of all cases of chronic pancreatitis in some countries [10], a recent report from India had only one case of autoimmune pancreatitis among 1,086 patients with chronic pancreatitis [31]. In the present report, we point out the fact that autoimmune pancreatitis may not be so rare inIndia. Early diagnosis of this potentially treatable disease is important because response to steroids is dramatic, and unnecessary surgical intervention with its attendant morbidity can be avoided.
Conflict of interest The authors have no potential conflict of interest
References
- Kazumori H, Ashizawa N, Moriyama N, Arima N, Hirakawa K, Adachi K, et al. Primary sclerosing pancreatitis and cholangitis. Int J Pancreatol 1998; 24:123-7. [PMID 9816546]
- Yoshida K, Toki F, Takeuchi T, Watanabe S, Shiratori K, Hayashi N. Chronic pancreatitis caused by an autoimmune abnormality.Proposal of the concept of autoimmune pancreatitis. Dig Dis Sci 1995; 40:1561-8. [PMID 7628283]
- Sarles H, Sarles JC, Muratore R, Guien C. Chronic inflammatory sclerosis of the pancreas--an autonomous pancreatic disease? Am J Dig Dis 1961; 6:688-98. [PMID 13746542]
- Sah RP, Pannala R, Chari ST, Sugumar A, Clain JE, Levy MJ, et al. Prevalence, diagnosis, and profile of autoimmune pancreatitis presenting with features of acute or chronic pancreatitis. Clin Gastroenterol Hepatol 2010; 8:91-6. [PMID 19800984]
- Chari ST, Smyrk TC, Levy MJ, Topazian MD, Takahashi N, Zhang L, et al. Diagnosis of autoimmune pancreatitis: the Mayo Clinic experience. Clin Gastroenterol Hepatol 2006; 4:1010-6. [PMID 16843735]
- Kamisawa T. IgG4-positive plasma cells specifically infiltrate various organs in autoimmune pancreatitis. Pancreas 2004; 29:167-8. [PMID 15257111]
- Frulloni L, Lunardi C, Simone R, Dolcino M, Scattolini C, Falconi M, et al. Identification of a novel antibody associated with autoimmune pancreatitis. N Engl J Med 2009; 361:2135-42. [PMID 19940298]
- Pearson RK, Longnecker DS, Chari ST, Smyrk TC, Okazaki K, Frulloni L, Cavallini G. Controversies in clinical pancreatology: autoimmune pancreatitis: does it exist? Pancreas 2003; 27:1-13. [PMID 12826899]
- Okazaki K, Kawa S, Kamisawa T, Naruse S, Tanaka S, Nishimori I, et al. Clinical diagnostic criteria for autoimmune pancreatitis: revised proposal. J Gastroenterol 2006; 41:626-31. [PMID 16932998]
- Otsuki M, Chung JB, Okazaki K, Kim MH, Kamisawa T, Kawa S, et al. Asian diagnostic criteria for autoimmune pancreatitis: consensus of Japan-Korea symposium on autoimmune pancreatitis. J Gastroenterol 2008; 43:403-8. [PMID 18600383]
- Ramachandran J, Chacko A, Peter S, Mathews J, Govil S. Autoimmune pancreatitis - An uncommon type of chronic pancreatitis. Indian J Gastroenterol 2004; 23:181-3. [PMID 15599002]
- Mancini G, CarbonaraA O, Heremsans JF Immunochemical quantitation of antigens by single radial immunodiffusion. Immunochemistry 1965; 2:235-54. [PMID 4956917]
- Tabata T, Kamisawa T, Takuma K, Anjiki H, Egawa N, Kurata M, et al. Serum IgG4 concentrations and IgG4 related sclerosing disease. Clin Chim Acta 2009; 408:25-8. [PMID 19580797]
- Wreesmann V, van Eijck CH, Naus DC, van Velthuysen ML, Jeekel J, Mooi WJ. Inflammatory pseudotumour (inflammatory myofibroblastictumour) of the pancreas: a report of six cases associated with obliterative phlebitis. Histopathology 2001; 38:105- 10. [PMID 11207823]
- Song Y, Liu QD, Zhou NX, Zhang WZ, Wang DJ. Diagnosis and management of autoimmune pancreatitis: experience from China. World J Gastroenterol 2008; 14:601-6. [PMID 18203294]
- Alexander S, Bourke MJ, Williams SJ, Bailey A, Gill A, Kench JG. Diagnosis of autoimmune pancreatitis with intraductal biliary biopsy and treatment of stricture with serial placement of multiple biliary stents.Gastrointest Endosc 2008; 68:396-9. [PMID 18279859]
- Ooi K, Merrett N. Autoimmune pancreatitis in a patient presenting with obstructive jaundice and pancreatic mass. HPB (Oxford) 2004; 6:126-7. [PMID 18333064]
- Wagle PK, Shetty GS, Tampi C. Lymphoplasmacyticsclerosing pancreatitis mimicking pancreatic cancer. Indian J Gastroenterol 2005; 24:86-7. [PMID 15879669]
- Kamisawa T, Egawa N, Nakajima H, Tsuruta K, Okamoto A, Kamata N. Clinical difficulties in the differentiation of autoimmune pancreatitis and pancreatic carcinoma. Am J Gastroenterol 2003; 98:2694-9. [PMID 14687819]
- Church NI, Pereira SP, Deheragoda MG, Sandanayake N, Amin Z, Lees WR, et al. Autoimmune pancreatitis: clinical and radiological features and objective response to steroid therapy in a UK series. Am J Gastroenterol 2007; 102:2417-25. [PMID 17894845]
- Ryu JK, Chung JB, Park SW, Lee JK, Lee KT, Lee WJ, et al. Review of 67 patients with autoimmune pancreatitis in Korea: a multicenter nationwide study. Pancreas 2008; 37:377-85. [PMID 18953249]
- Deshpande V, Mino-Kenudson M, Brugge WR, Pitman MB, Fernandez-del Castillo C, Warshaw AL, Lauwers GY. Endoscopic ultrasound guided fine needle aspiration biopsy of autoimmune pancreatitis, diagnostic criteria and pitfalls. Am J Surg Pathol 2005; 29:1464-71. [PMID 16224213]
- Chari ST, Takahashi N, Levy MJ, Smyrk TC, Clain JE, Pearson RK, et al. A diagnostic strategy to distinguish autoimmune pancreatitis from pancreatic cancer.Clin Gastroenterol Hepatol 2009; 7:1097-103. [PMID 19410017]
- Kawamoto S, Siegelman SS, Hruban RH, Fishman EK. Lymphoplasmacyticsclerosing pancreatitis (autoimmune pancreatitis): evaluation with multidetector CT. Radiographics 2008; 28:157-70. [PMID 18203936]
- Sahani DV, Kalva SP, Farrell J, Maher MM, Saini S, Mueller PR, et al. Autoimmune pancreatitis: imaging features. Radiology 2004; 233:345-52. [PMID 15459324]
- Takahashi N, Kawashima A, Fletcher JG, Chari ST. Renal involvement in patients with autoimmune pancreatitis: CT and MR imaging findings. Radiology 2007; 242:791-801. [PMID 17229877]
- Kim KP, Kim MH, Song MH, Lee SS, Seo DW, Lee SK. Autoimmune chronic pancreatitis. Am J Gastroenterol 2004; 99:1605-16. [PMID 15307882]
- Takikawa H, Manabe T. Primary sclerosing cholangitis in Japananalysis of 192 cases. J Gastroenterol 1997; 32:134-7. [PMID 9058310]
- Kubota K, Kato S, Akiyama T, Yoneda M, Fujita K, Ogawa M, et al. Differentiating sclerosing cholangitis caused by autoimmune pancreatitis and primary sclerosing cholangitis according to endoscopic duodenal papillary features. Gastrointest Endosc 2008; 68:1204-8. [PMID 19028233]
- Kamisawa T, Takuma K, Anjiki H, Egawa N, Kurata M, Honda G, Tsuruta K. Sclerosing cholangitis associated with autoimmune pancreatitis differs from primary sclerosing cholangitis. World J Gastroenterol 2009; 15:2357-60. [PMID 19452578]
- Balakrishnan V, Unnikrishnan AG, Thomas V, Choudhuri G, Veeraraju P, Singh SP, et al. A prospective nationwide study of 1,086 subjects from India.JOP. J Pancreas (Online) 2008; 9:593-600. [PMID 18762690]




