Keywords
Cell Differentiation; Insulin; Islets of Langerhans; Pancreas, Exocrine
Abbreviations
BSA: bovine serum albumin; DAPI: diamindino 2 phenylindole; DMEM: Dulbecco's Modified Eagle Medium; DMEM/F-12: Dulbecco's Modified Eagle Medium-F12; FCS: fetal calf serum; HGF: hepatocyte growth factor; HBSS: Hank’s balanced salt solution; ITS: insulin transferrin-selenium; PBS: phosphate buffered saline
INTRODUCTION
Replacement of the damaged pancreatic beta cells in insulin-dependent diabetic patients represents a strategy for restoring glycemic control. Among others, the most severe limitations of this approach are the scarcity of organ donation with respect to the large number of diabetic patients. New strategies for obtaining insulin producing cells from other sources are under extensive investigation. Beta cells in the adult pancreas have a brief life span after which they undergo apoptosis [1]. It has been documented that senescent beta cells are replaced by the replication of existing mature beta cells [2] and also by the differentiation or neogenesis of beta cells deriving from exocrine or intra-islet pancreatic precursor cells [3, 4, 5]. The exact nature of pancreatic stem cells is still not well defined [6] but precursor cells have been shown to be present either in the exocrine tissue or within the islets [3]. There is clear evidence of the presence of pancreatic progenitor cells in the ductal epithelium of the pancreas [7]. Thus, cultures of mouse pancreatic epithelium under appropriate conditions resulted in cell differentiation with insulin secretion in a glucose-dependent manner. These cells have been shown to normalize hyperglycemia after implantation into diabetic mice [8]. Similar results were reported for human pancreatic epithelial cells which have been differentiated by specific factors and matrigel culture forming three dimensional islet-like clusters [9].
It has been suggested that progenitor cells in the rat and human pancreas are positive for nestin [10, 11, 12, 13]. Nestin-expressing cells are also located within the pancreatic ducts as undifferentiated cells which are distinct from ductal epithelial cells. All these studies used several differentiation factors to induce the expansion of pancreatic islet cells in vitro and/or in vivo, including hepatocyte growth factor (HGF), glucagon-like peptide- 1, parathyroid hormone-related protein, and others [14, 15, 16]. In addition, several studies have demonstrated that HGF is an insulinotropic factor for adult islet beta cells [17, 18] and it is also able to induce the expression of insulin in a rat pancreatic cell line which normally lacks expression of this hormone [19]. All this evidence suggests that the in vitro expansion and culture of the progenitors of beta cells may represent an alternative to the use of differentiated beta cells from donor pancreata.
The aim of our study was to assess whether the in vitro differentiation of islet and exocrine tissue may effectively result in the production of insulin-containing cells, and to quantify the yield of this process.
MATERIALS AND METHODS
Experimental Design
Bovine exocrine tissue (n=4) and islets (n=4) were cultured in DMEM with serum. The proliferating capacity of both exocrine and islet tissues were analyzed by cell counting after 4 weeks of DMEM culture. In addition, the fraction of the volume of the cell clusters occupied by insulin-containing cells after differentiation was estimated by morphometrical analysis of the histochemically identified cells, and immunohistochemical data were confirmed by semi-quantitative RTPCR results evaluated at passages 0 and 3.
Exocrine and Islet Tissue Separation
Exocrine and islet tissues were obtained from bovine pancreas digestion with collagenase using a modification of the automated method [20] and were purified by centrifugation on a discontinuous gradient. Briefly, the main pancreatic duct was cannulated and perfused with collagenase solution (1.8 U/mL, type P, Roche Diagnostic, Mannheim, Germany). After ductal distension, the splenic lobe of the pancreas was placed into a perfusion chamber. Recirculating collagenase solution in a continuous digestion apparatus was warmed to 37°C throughout the dissociation of the pancreas. Effluent samples from the perfusion chamber were evaluated during digestion to determine the stage of the digestion process. When the majority of islets appeared free from exocrine tissue, the dissociation chamber was flushed with cold (4°C) Hanks’ balanced salt solution (HBSS, Gibco InVitrogen Co., Paisley, Scotland, United Kingdom), at 400 mL/min supplemented with 1% of fetal calf serum (FCS, Gibco InVitrogen Co., Paisley, Scotland, United Kingdom). The digested tissue was collected and purified by centrifugation on a Histopaque gradient (1.077 g/mL, Sigma, St. Louise, MO, U.S.A.). Islet fraction was recovered on the surface of the Histopaque layer, while the denser bovine acinar fraction was recovered in the pellet. After three washings with phosphate buffer saline, the equivalent of 80 μL packed exocrine pellets, or 5,000 islets, were placed in culture-treated plastic dishes (Falcon, Becton Dickinson, Meylan Cedex, France). The cells were cultured at 37°C in a humidified atmosphere (5% CO2) in DMEM medium (Sigma, St. Louise, MO, U.S.A.) containing 10% FCS, 100 U/mL penicillin, 100 μg/mL streptomycin (Sigma, St. Louise, MO, U.S.A.) and 50 μg/mL geneticin (Gibco InVitrogen Co., Paisley, Scotland, United Kingdom).
Culture Conditions
After having been cultured for 3-4 days, the non-adherent tissue was removed and the adherent cells were expanded for up to one week with a change of medium every 2-3 days. After one week, when the adherent cells were confluent in a monolayer, the cells were detached from the dishes by trypsin digestion and expanded on tissue culture dishes. The trypsinized cells were separated into different culture conditions to favor the proliferation or differentiation of the cells. The medium used for proliferation was DMEM containing 10% FCS and 100 U/mL penicillin, 100 μg/mL streptomycin. To inhibit the growth of fibroblasts, a low concentration of antibiotic geneticin (50 μg/mL) was added to the culture medium [21, 22]. The medium used to stimulate differentiation was DMEM/F12 (16.7 mM glucose, Sigma, St. Louise, MO, U.S.A.) with 1 g/L insulin-transferrinselenium (ITS) supplement (5 mg/L insulin + 5 mg/L transferrin + 5 mg/L selenium, Sigma, St. Louise, MO, U.S.A.), 100 U/mL penicillin, 100 μg/mL streptomycin, 2 g/L bovine serum albumin (BSA, Sigma, St. Louise, MO, U.S.A.), 10 mM nicotinamide (Sigma, St. Louise, MO, U.S.A.), and 10 ng/mL HGF (Sigma, St. Louise, MO, U.S.A.). Cells were maintained in culture for one month.
Proliferation Assay
Cell proliferation was evaluated with a Burker chamber by counting the cells at different times during in vitro culture. The cells were removed from the culture dish by treatment with trypsin-EDTA (Gibco InVitrogen Co., Paisley, Scotland, United Kingdom) and were resuspended in medium containing serum. Aliquots of approximately 10 μL were used for counting in triplicate. The procedure was performed twice a week for about one month.
Immunofluorescence
The expression of epithelial- and nestinspecific markers was investigated by immunofluorescence analysis. Briefly, the sections were stained with mouse monoclonal anti-keratin 7,17 antibody (Chemicon Int, Temecula, CA, U.S.A.), or mouse monoclonal anti-nestin antibody (Chemicon Int, Temecula, CA, U.S.A.). Coverslips with cell monolayers were fixed in 2% paraformaldheyde plus 2% saccarose for 10 min at room temperature. Sections with nonspecific binding of antibodies were incubated for 10 min at room temperature with blocking solution (3% BSA and 10% fetal calf serum in PBS). Monolayer cultures were incubated for 3 min in 0.1% Triton X-100 before primary antibody incubation. The sections were then incubated with primary antibodies for 2 h at room temperature (keratin 1:100, nestin 1:100). After washing in PBS, the sections stained for keratin and nestin were incubated with Cy3 conjugated donkey antimouse IgG antibody (Jackson Immunoresearch, West Grave, PA, U.S.A.), or fluorescein isothiocyanate (FITC) conjugate donkey anti-mouse IgG (Sigma, St. Louise, MO, U.S.A.) for 1 h at room temperature. Counterstaining with diamindino 2 phenylindole (DAPI) (1 μg/mL, Sigma, St. Louise, MO, U.S.A.) for 5 min at room temperature was performed to label cell nuclei. After final washings in PBS, all slides were mounted and examined by laser confocal microscopy (LSM 510 Meta, Zeiss, Jena, Germany).
Immunohistochemistry
For identification of the insulin-containing cells, immunohistochemical analysis was performed by fixation of islet-like structures in Bouin’s solution. After a 4 h fixation, the samples were paraffin-embedded using previously described methods [23]. Briefly, 3 μm thin sections were collected and processed for immunohistochemical detection of insulin using the alkaline phosphate-fast red technique. Slides were blocked in PBS (plus 1% BSA) for 30 min at room temperature and stained using mouse antiinsulin (diluted 1:100, Sigma, St. Louise, MO, U.S.A.) for 2 h at room temperature. The sections were then washed and incubated with horse biotinylated anti mouse antibody (Vector, Burlingame, CA, U.S.A.) for 30 minutes. The slides were then washed and incubated with alkaline phosphataseconjugated streptavidin (Boerhinger, Mannheim, Germany) for 30 min, followed by washings and development with Fast Red substrate (Boerhinger, Mannheim, Germany). The sections were counterstained with Harristype hematoxylin and mounted using aqueous mounting medium (Bio-Optica, Milan, Italy). PBS (pH 7.4) was used for all antibody dilutions and washings.
Morphometrical Analysis of Insulin Containing Cells
Sections of islet-like structures or pancreatic islets stained for insulin were used for morphometrical analysis of the insulincontaining cells. Sections were examined at light microscopy (Zeiss, Jena, Germany) and images were acquired using a computer-based image analysis system. For each slide, the islet-like structures or pancreatic islets encountered while moving the microscope stage with an S-shaped path were digitized using a 40x objective. The volume density (Vv) of the tissue reacting with insulin antibody was estimated by point counting using an orthogonal grid digitally overlaid on the stained section image (Image J v. 1.31, National Institute of Mental Health, Bethesda, Maryland, U.S.A.). For each section, the number of grid points hitting the positive staining and total number of grid points hitting the reference tissue were counted. Volume density was calculated as the percentage ratio between grid points in the positive areas over total points in the islet-like structures or pancreatic islets [24].
mRNA Extraction and Real-Time Quantitative PCR
Total RNA was extracted from the islets and from the three-dimensional aggregates in the presence of TRIzol reagent (Invitrogen, Carlsbad, CA, U.S.A.). DNase treated RNA was primed with random hexamers and reverse transcribed to cDNA using Superscript II Reverse Transcriptase (Invitrogen, Carlsbad, CA, U.S.A.). Quantitative real-time PCR was performed utilizing a TaqMan AB Prism 5700 Sequence Detection System (Applied Biosystems, Foster City, CA, U.S.A.) with SYBR green PCR core reagents. To amplify the bovine insulin transcripts, the following primers based on Genbank sequence NM_173926.1 were used: For (300 nM) 5’-GCA GAA GCG TGG CAT CGT-3’; Rev (300 nM) 5’-GGG CAG GCC TAG TTA CAG TAG TTC-3’. Genomic DNA was not amplified since primers spanned an exon junction. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) served as a housekeeping gene to assess the overall cDNA content. GAPDH primers were as follows: For (300 nM) 5’- GGC ATC GTG GAG GGA CTT ATG-3’; Rev (300 nM) 5’-GGG CCA TCC ACA GTC TTC TG-3’. After an initial holding step of 2 min at 50°C and 10 min at 95°C, samples were cycled 40 times at 95°C for 15 sec and at 60°C for 60 sec. All reactions were carried out in triplicate. Similar amplification efficiencies were demonstrated for both the target and the housekeeping genes by analyzing serial cDNA dilutions, showing an absolute value of the slope of log cDNA amount versus Δ threshold cycle (Ct) (Ct target-Ct housekeeping gene) of less than 0.1. Data were calculated relative to the control pancreatic islets using the ΔΔCt method and GAPDH as reference gene.
STATISTICS
Data are reported as mean±SE. Data were analyzed by mean of the paired and unpaired Student t test by using the SPSS Version 13.0 for Windows (SPSS, Chicago, IL, U.S.A.). Two-tailed P values less than 0.05 were considered statistically significant.
RESULTS
Suspensions of exocrine and islet tissue obtained after the enzymatic digestion and purification of the bovine pancreas were cultured in DMEM supplemented with 10% FCS. The majority of the exocrine tissue adhered to the plate after a few days of being cultured, and cells grew out of the exocrine tissue. In islet culture, after attachment to the plastic surface, the islets lost their threedimensional architecture and a cell monolayer grew radially from the islets. After 3-4 days, the non-adherent cells were removed while the attached cells were left to proliferate and reached confluence in about one week (Figure 1). Upon reaching confluence, the cells were detached and re-seeded at a lower density. After the first passages in culture, exocrine tissue cells grew forming a monolayer with a prevalent cuboid morphological appearance. As shown in Figure 2a, most of these cells were positive for cytokeratins 7,17, a specific marker of epithelial cells. Cells positive for nestin in this monolayer were rare (Figure 2c). In a monolayer of cells derived from islet tissue, fibroblast-like morphology was predominant and these cells were identified as nestin positive by immunofluorescence analysis (Figure 2b). Nestin-positive cells were more prevalent in the islet-derived monolayer than in the exocrine derived cell population examined.
Figure 1. Phase contrast of exocrine tissue (panels a and c) and islet tissue (panels b and d), after a 1 week culture in
DMEM low glucose. Original magnification: 20x (a and b) and 40x (c and d).
Figure 2. Confocal microscopy of exocrine tissue
immunofluorescence for keratin 7,17 (panel a) and for
nestin (panel c) and islet tissue immunofluorescence
for nestin (panel b). Nuclei are stained in blue with
DAPI. Original magnification: 60x.
After the first passage, both exocrine- and islet-derived cells divided rapidly and became confluent within 7 days. We analyzed the proliferating capacity of both exocrine and islet tissues by cell counting. The growth curve estimations are shown in Figure 3. After 4 weeks in culture, the number of cells from the exocrine tissue showed a 69.5±10.0 fold increase (n=4, P=0.006, P=0.007, and P=0.007 vs. 1, 2, and 3 weeks, respectively) with respect to the number of cells in the starting monolayer. At the same time, the cells originating from the islet tissue showed a 31.2±11.4 fold increase (n=3, P=0.118, P=0.191, and P=0.261 vs. 1, 2 and 3 weeks, respectively). The comparison between exocrine and islet tissues showed a significant difference after 2 weeks (P=0.012) while the difference after 4 week was of a borderline significant level (P=0.053).
Figure 3. Proliferation of cell monolayers from
exocrine (n=4) and islet (n=3) cells during a 4 week
culture in DMEM medium (mean±SE).
To induce differentiation of the proliferating cells into insulin containing ones, the cells were treated with growth factors known to induce beta cell differentiation. Actually, after trypsinization, the culture conditions of a fraction of the proliferating cells were modified by changing the cell medium from DMEM with 5.5 mM glucose to DMEM/F-12 with 16.7 mM glucose plus ITS, HGF, and nicotinamide. After this change of medium, cells from both the exocrine and the islet tissue began to aggregate into spherical cell clusters. These three-dimensional cell clusters remained attached to the plastic substrate for a period of 10-12 days and then subsequently detached from the surface and remained floating in the medium. Islet-like structures from exocrine and islet tissue are shown in Figures 4a and 4b, respectively. They were similar in size and morphology to the pancreatic islets shown in Figure 4c. Immunohistochemical analysis of these structures with anti-insulin antibody showed the presence of some positive cells in both exocrine- and islet-derived structures, as shown in Figure 5.
Figure 4. Phase contrast of islet-like structures derived
from exocrine tissue (panel a) and islet tissue (panel b). Pancreatic islets are reported for comparison (panel c). Original magnification: 60x.
Figure 5. Immunohistochemical staining for insulin of
islet-like structures derived from exocrine tissue (panel a) and islet tissue (panel b). Pancreatic islets are
reported for comparison (panel c).
We estimated the fraction of the volume of the cell clusters occupied by the insulincontaining cells using morphometrical analysis of histochemically identified cells. The volume density of the cells positive for anti-insulin antibody was 0.47±0.11% and 6.73±2.70% for islet-like structures derived from exocrine and from islet tissue, respectively (n=4, P=0.059) while the same volume density measured in pancreatic islets samples averaged 82.2±1.86% (n=3; P<0.001 vs. both exocrine and islet tissues).
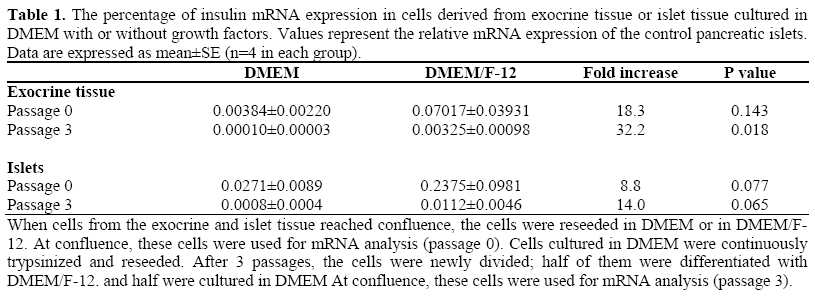
Immunohistochemical data were confirmed by semi-quantitative RT-PCR results. This analysis showed that cells grown in DMEM had very low levels of insulin mRNA. We observed 0.004±0.002% and 0.027±0.009% mRNA insulin in exocrine tissue and in islet tissue, respectively. After differentiation, insulin expression in islet-like structures increased. As shown in Table 1, the increase of insulin expression was 18.3 fold in isletlike structures obtained from exocrine tissue (0.070±0.039% mRNA insulin) and 8.8 fold in islet-like structures obtained from islet tissue (0.237±0.098% mRNA insulin) as compared to the gene expression of the undifferentiated cells to which the arbitrary unit value was assigned. The increase of insulin mRNA was enhanced with passages in culture for both cells derived from islets and from exocrine preparations.
DISCUSSION
In the present study, we initially quantified the proliferation capacity of bovine exocrine and islet pancreatic tissue in vitro, and then the differentiation of expanded monolayers in insulin containing islet-like structures. When cultured in DMEM medium, both bovine pancreatic exocrine and islet cells adhere to the plastic surface of the culture dishes forming a monolayer of cells with different phenotypes. Thus, cuboidal cells were predominant in exocrine rather than in isletpurified preparations and these cells are likely ductal epithelial cells. This is confirmed by positive immunofluorescence staining of these cells for cytokeratins (Figure 2). In islet-derived cell monolayers, we observed the presence of two distinct types of cells, fibroblast-like cells and rounded cells which derived from the breakup of the islets when they attached to the plastic surface. We characterized the fibroblast-like cells as nestin-positive cells (Figure 3). Zulewsky et al. [13] suggested that these cells are a distinct population which resides within pancreatic islets and may participate in the neogenesis of endocrine cells. Moreover, the presence of these cells was described in exocrine tissue cultures by Street et al. who characterized the expression and localization of nestin in the human acinar pancreas [25]. Our present data show that cells from exocrine and islet tissue cultured in the presence of ITS, HGF, and nicotinamide aggregate from the monolayer and form isletlike structures. On the contrary, the absence of growth factors favors the formation of a monolayer of cells which reaches confluence in 2-4 days. The ability of forming a cell monolayer is also maintained after trypsinization by these cells. We found that exocrine and islet cells rapidly proliferate giving rise to a population of cells which can subsequently differentiate into cells with a phenotype suggestive of pancreatic beta cells, as shown by the presence of insulin-positive cells in these three-dimensional cell clusters. Among the factors used to differentiate pancreatic cells, HGF is known to stimulate the proliferation of keratinocytes, kidney tubular epithelial cells and endothelial cells [26]. In addition to its mitogenic activity, HGF also has motogenic [27] and morphogenetic activity [28] and has previously been shown to be a potent regulator of beta cell phenotype [18, 20]. Another factor present in DMEM/F-12 media is nicotinamide which has been shown to protect islets from inflammation and to stimulate endocrine cell differentiation of beta cells in vivo [29]. Otonkoski et al. [30] showed that the morphological and functional differentiation of human pancreatic beta cells could be induced by nicotinamide the human fetal pancreas cultures.
The observation that culturing cells in selected differentiating media induced the presence of insulin-containing cells cannot be taken as a demonstration that these differentiated cells produce the hormone. Actually, as pointed out by Sipione et al. [31] in the case of embryonic stem cells, insulin immunoreactivity can be due to insulin uptake from the medium rather than to endogenously synthesized insulin. To verify whether our positively-stained cells effectively produced insulin or only stored insulin from the culture media, we estimated insulin mRNA expression. Thus, insulin production by these cells as a consequence of glucose challenge would be undetectable since the number of insulin-positive cells was very low. Insulin mRNA expression in differentiated cells using RT-PCR suggests that insulin is indeed produced rather than uptaken from the culture medium. In particular, in line with insulin expression, however, insulin mRNA expression was also very low. RT-PCR performed after having been cultured for one week in DMEM medium showed a very low content of insulin mRNA both in exocrine- and islet-derived cell monolayers. The endocrine contamination eventually present in the exocrine preparation was almost completely lost after a few days in culture. Similarly, also in islet-derived cells, after one week in culture, the ability to produce insulin is completely lost. When exposed to differentiating medium, with the addition of growth factors, few cells differentiate into insulin-containing cells. In comparison with the starting monolayer, we measured a significant increase in insulin mRNA in isletlike structures derived both from exocrine and islet tissue, suggesting that, under these conditions, some cells differentiated from precursor cells present in the adult bovine pancreas. However, as shown by the amount of insulin mRNA expression in comparison to that expressed by control islets (Table 1), the quantity of cells expressing insulin mRNA was very low. Even at the first passage, insulin mRNA expression was less than 0.1 and 0.3% of that of normal islets for differentiated cells from exocrine and islet tissue, respectively. The same conclusion can be drawn from morphometrical data which showed that the volume density of insulincontaining cells was less than 0.4% for exocrine tissue. An higher expression was observed in islet tissue where the volume density of insulin-containing cells was about 7% but still very low in comparison to that of normal islets (greater than 80%).
Altogether, these data demonstrated that it is possible to induce bovine pancreatic cell proliferation and differentiation in vitro, but the efficiency of this process is very low and not comparable to that shown by beta cell mass used in experimental and clinical islet transplantation. One could speculate that the selection of progenitor cells, isolated and selectively expanded from the starting cell population, may improve beta cell differentiation yield. However, the exact procedure for performing this cell selection is still not known and is worth investigating in the attempt to regenerate in vitro cell populations suitable for pancreatic beta cell function replacement.
Acknowledgements
We thank Tiziana Plati for excellent her technical assistance during islet isolation. Part of this study was supported by a research grant from “Fondazione Cariplo” (contract number 2002.2310/10.496)
References
- Finegood DT, Scaglia L, Bonner-Weir S. Dynamics of beta-cell mass in the growing rat pancreas. Estimation with a simple mathematical model. Diabetes 1995; 44:249-56. [PMID 7883109]
- Dor Y, Brown J, Martinez OI, Melton DA. Adult pancreatic beta-cells are formed by self-duplication rather than stem-cell differentiation. Nature 2004; 429:41-6. [PMID 15129273]
- Lock LT, Tzanakakis ES. Stem/Progenitor cell sources of insulin-producing cells for the treatment of diabetes. Tissue Eng 2007; 13:1399-412. [PMID 17550339]
- Baeyens L, De Breuck S, Lardon J, Mfopou JK, Rooman I, Bouwens L. In vitro generation of insulinproducing beta cells from adult exocrine pancreatic cells. Diabetologia 2005; 48:49-57. [PMID 15616797]
- Gallo R, Gambelli F, Gava B, Sasdelli F, Tellone V, Masini M, et al. Generation and expansion of multipotentmesenchymal progenitor cells from cultured human pancreatic islets. Cell Death Differ 2007; 14:1-12. [PMID 17612586]
- Limbert C, Path G, Jakob F, Seufert J. Beta-cell replacement and regeneration: Strategies of cell-based therapy for type 1 diabetes mellitus. Diabetes Res Clin Pract 2008; 79:389-99. [PMID 17854943]
- Bonner-Weir S, Sharma A. Pancreatic stem cells. J Pathol 2002; 197:519-26. [PMID 12115867]
- Ramiya VK, Maraist M, Arfors KE, Schatz DA, Peck AB, Cornelius JG. Reversal of insulin-dependent diabetes using islets generated in vitro from pancreatic stem cells. Nat Med 2000; 6:278-82. [PMID 10700229]
- Bonner-Weir S. beta-cell turnover: its assessment and implications. Diabetes 2001; 50 Suppl 1:S20-4. [PMID 11272192]
- Eberhardt M, Salmon P, von Mach MA, Hengstler JG, Brulport M, Linscheid P, et al. Multipotentialnestin and Isl-1 positive mesenchymal stem cells isolated from human pancreatic islets. BiochemBiophys Res Commun 2006; 345:1167-76. [PMID 16713999]
- Ueno H, Yamada Y, Watanabe R, Mukai E, Hosokawa M, Takahashi A, et al. Nestin-positive cells in adult pancreas express amylase and endocrine precursor Cells. Pancreas 2005; 31:126-31. [PMID 16024998]
- Esni F, Stoffers DA, Takeuchi T, Leach SD. Origin of exocrine pancreatic cells from nestin-positive precursors in developing mouse pancreas. Mech Dev 2004; 121:15-25. [PMID 14706696]
- Zulewski H, Abraham EJ, Gerlach MJ, Daniel PB, Moritz W, Müller B, et al. Multipotentialnestinpositive stem cells isolated from adult pancreatic islets differentiate ex vivo into pancreatic endocrine, exocrine, and hepatic phenotypes. Diabetes 2001; 50:521-33. [PMID 11246871]
- Abraham EJ, Leech CA, Lin JC, Zulewski H, Habener JF. Insulinotropic hormone glucagon-like peptide-1 differentiation of human pancreatic isletderived progenitor cells into insulin-producing cells. Endocrinology 2002; 143:3152-61. [PMID 12130581]
- Garcia-Ocaña A, Takane KK, Syed MA, Philbrick WM, Vasavada RC, Stewart AF. Hepatocyte growth factor overexpression in the islet of transgenic mice increases beta cell proliferation, enhances islet mass, and induces mild hypoglycemia. J Biol Chem 2000; 275:1226-32. [PMID 10625667]
- Porter SE, Sorenson RL, Dann P, Garcia-Ocana A, Stewart AF, Vasavada RC. Progressive pancreatic islet hyperplasia in the islet-targeted, parathyroid hormonerelated protein-overexpressing mouse. Endocrinology 1998; 139:3743-51. [PMID 9724026]
- Otonkoski T, Cirulli V, Beattie M, Mally MI, Soto G, Rubin JS, Hayek A. A role for hepatocyte growth factor/scatter factor in fetal mesenchyme-induced pancreatic beta-cell growth. Endocrinology 1996; 137:3131-9. [PMID 8770939]
- Otonkoski T, Beattie GM, Rubin JS, Lopez AD, Baird A, Hayek A. Hepatocyte growth factor/scatter factor has insulinotropic activity in human fetal pancreatic cells. Diabetes 1994; 43:947-53. [PMID 8013761]
- Furukawa M, Zhang YQ, Nie L, Shibata H, Kojima I. Role of mitogen-activated protein kinase and phosphoinositide 3-kinase in the differentiation of rat pancreatic AR42J cells induced by hepatocyte growth factor. Diabetologia 1999; 42: 450-6. [PMID 10382592]
- Ricordi C, Lacy PE, Finke EH, Olack BJ, Scharp DW. Automated method for isolation of human pancreatic islets. Diabetes 1988; 37:413-20. [PMID 3288530]
- Inagaki T, Matsuwari S, Shimada K, Abe N, Takahashi R, Maeda S. Effective removal of the contaminating host fibroblasts for establishment of human-tumor cultured lines. Hum Cell 1993; 6:137-42. [PMID 8217952]
- Halaban R, Alfano FD. Selective elimination of fibroblasts from cultures of normal human melanocytes. In Vitro 1984; 20:447-50. [PMID 6724622]
- Remuzzi A, Puntorieri S, Alfano M, Macconi D, Abbate M, Bertani T, Remuzzi G. Pathophysiologic implications of proteinuria in a rat model of progressive glomerular injury. Lab Invest 1992 ; 67:572-9. [PMID 1434536]
- Weibel ER. Stereological methods. Practical Methods for Biological Morphometry. Academic Press, London 1979.
- Street CN, Lakey JR, Seeberger K, Helms L, Rajotte RV, Shapiro AM, Korbutt GS. Heterogenous expression of nestin in human pancreatic tissue precludes its use as an islet precursor marker. J Endocrinol 2004; 180:213-25. [PMID 14765974]
- Wang TC, Bonner-Weir S, Oates PS, Chulak M, Simon B, Merlino GT, et al. Pancreatic gastrin stimulates islet differentiation of transforming growth factor alpha-induced ductular precursor cells. J Clin Invest 1993; 92:1349-56. [PMID 8376589]
- Uehara Y, Kitamura N. Expression of a human hepatocyte growth factor/scatter factor cDNA in MDCK epithelial cells influences cell morphology, motility, and anchorage-independent growth. J Cell Biol 1992; 117:889-94. [PMID 1315783]
- Montesano R, Matsumoto K, Nakamura T, Orci L. Identification of a fibroblast-derived epithelial morphogen as hepatocyte growth factor. Cell 1991; 67:901-8. [PMID 1835669]
- Vague P, Picq R, Bernal M, Lassmann-Vague V, Vialettes B. Effect of nicotinamide treatment on the residual insulin secretion in type 1 (insulin-dependent) diabetic patients. Diabetologia 1989; 32:316-21. [PMID 2526769]
- Otonkoski T, Beattie GM, Mally MI, Ricordi C, Hayek A. Nicotinamide is a potent inducer of endocrine differentiation in cultured human fetal pancreatic cells. J Clin Invest 1993; 92:1459-66. [PMID 8104197]
- Sipione S, Eshpeter A, Lyon JG, Korbutt GS, Bleackley RC. Insulin expressing cells from differentiated embryonic stem cells are not beta cells. Diabetologia 2004; 47:499-508. [PMID 14968299]






