Keywords
Hyperinsulinism; Hyperplasia; Hypoglycemia; Insulinoma; Islets of Langerhans
INTRODUCTION
The vast majority of cases of hyperinsulinemic hypoglycemia result from insulinoma or exogenous insulin administration. However, rare cases evolve in the setting of diffuse islet cell hyperplasia arising from the ductal epithelium, a condition known as nesidioblastosis. Seventy-three cases of this entity have been described in the English literature since 1938, when the disorder was first defined by Laidlaw [1]. The vast majority of these cases have been documented in children, with only rare cases seen in adults.
The diagnosis of this disorder is complex, as traditional imaging studies are not revealing, and historical questioning of the patient may reveal a complicated and confusing pattern of symptoms. The fundamental physiologic process underlying the condition involves insulin hypersecretion leading to significant hypoglycemia. In light of this, the technique of selective arterial calcium stimulation with hepatic venous sampling has been utilized in some clinical centers to aid diagnosis [2, 3]. We report a rare case of adult nesidioblastosis in a 45-year-old woman who presented with hypoglycemia and seizures. The selective arterial calcium stimulation with hepatic venous sampling procedure was utilized to facilitate diagnosis and guide surgical intervention, and the subsequent resection specimen confirmed the diagnosis of nesidioblastosis.
CASE REPORT
A 45-year-old woman presented to the emergency department with a history of lethargy and new-onset tonic-clonic seizures. Blood glucose measurement on presentation was noted to be 10 mg/dL (reference range: 70-100 mg/dL). The patient, whose only significant past medical history was that of oxycodone abuse, had experienced similar lethargic episodes on six occasions over the preceding eighteen months but never had developed frank seizure activity. The patient’s medications at presentation included methadone for oxycodone abuse and alprazolam, which she occasionally used for anxiety. On admission, initial treatment included intravenous (i.v.) infusion of 5% dextrose in water. After volume and glycemic resuscitation, the patient’s glucose increased to 67 mg/dL. However, within three hours, the patient’s glucose again decreased to 37 mg/dL despite the dextrose infusion. As such, a peripherally inserted central catheter line was immediately placed and the patient was aggressively treated with continuous infusion of 20% dextrose in water.
In addition to basic chemistry assessment, the patient underwent immediate urine toxicology testing which was negative for barbiturates, salicylates, ethanol, acetaminophen, and multiple tricyclic anti-depressants on admission. Based on the patient’s occupation as a medical health professional, and given her past substance abuse, factitious hypoglycemia was high on the admission differential diagnosis. Suspicion was increased when the patient mentioned that several of her clients were being treated with oral hypoglycemic medications and that she had regular access to these drugs. As such, the patient was admitted, placed on continuous 24-hour observation, and underwent screening for the presence of the following sulfonylureas in urine by high performance liquid chromatography: glipizide, glyburide, tolazamide, and tolbutamide. Testing on two separate specimens collected over 48 hours was negative for the above tested medications. The patient remained persistently hypoglycemic during this period even in the face of continuous i.v. dextrose infusion.
While observation and testing for factitious hypoglycemia was on-going, several additional laboratory studies were performed. Baseline endocrine function tests revealed: thyroid-stimulating hormone 0.5 μIU/mL (reference range: 0.3-4.2 μIU/mL), prolactin 19 ng/mL (reference range: 0-20 ng/mL), intact parathyroid hormone 20 pg/mL (reference range: 6-40 pg/mL), and morning cortisol 12 μg/dL (reference range: 7-25 μg/dL). Serial insulin testing by solid-phase chemiluminescent assay (Immulite 2000, Siemens Medical Diagnostics, Los Angeles, CA, USA) was performed over the first week of admission and revealed hyperinsulinemic hypoglycemia, exemplified by insulin levels of 54 μU/mL (reference range: 0-15 μU/mL) in association with blood glucose of 25 mg/dL on day 6 of admission [4]. Additionally, Cpeptide levels drawn on day 2 of admission revealed levels of 4.5 mg/mL (reference range: 0.9-4.3 mg/mL) while proinsulin was 11.2 pmol/mL (reference range: 0-8.8 pmol/mL). Testing for anti-insulin antibodies was negative.
This endocrine testing, in conjunction with the negative oral hypoglycemic toxicology results, was most suggestive of a diagnosis of insulinoma, particularly as the patient remained continuously hypoglycemic through the first week of her admission. However, the patient’s subsequent diagnostic imaging studies failed to reveal a pancreatic mass lesion or any other abnormalities. Endoscopic ultrasound demonstrated a normal pancreatic duct, a well-circumscribed, rounded, hypoechoic area measuring 6 mm adjacent to the spleen along the pancreatic tail, and a 4 mm oblong hypoechoic area at the junction of the pancreatic neck and body; all findings were considered non-specific and inconclusive. An octreotide scan was also performed with negative results.
With the patient remaining persistently hypoglycemic, even with the continuous administration of up to 200 mL/h of i.v. 20% dextrose in water, the primary clinical team opted for more invasive diagnostic testing in an attempt to locate a presumed insulin secreting tumor in the pancreas. At this time, the patient was referred to the interventional radiology service for selective arterial calcium stimulation with hepatic venous sampling. In this procedure, arteries supplying the pancreas are cannulated with the subsequent injection of intra-arterial calcium gluconate, a pancreatic secretagogue which stimulates insulin release [3]. Serial venous whole blood samples are then collected from the right hepatic veins which are tested for insulin levels pre-stimulation and at 30, 60, and 120 seconds post-stimulation. For this study, a two-fold or higher increase in insulin above baseline (pre-calcium) levels is suggestive of hyperactive beta-cell activity. For our patient, branches of the superior mesenteric, right hepatic, mid-splenic, proximal splenic, proper hepatic and gastroduodenal arteries were injected with calcium gluconate, and multiple consecutive venous whole blood samples were extracted from the right hepatic vein for assessment of insulin. All regions of the pancreas, except those supplied by the superior mesenteric artery, showed large increases above baseline insulin levels with calcium gluconate stimulation (Table 1). In fact, the insulin levels were somewhat greater than expected in the absence of metastatic disease to the liver. However, as stated previously, imaging studies were clearly negative in this regard. We have no plausible explanation for this finding, although it is not ultimately inconsistent with the clinicopathologic picture.
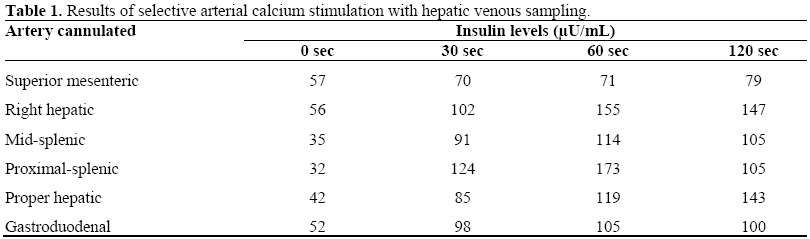
Based on the near-global increase in pancreatic beta-cell activity seen during selective arterial calcium stimulation with hepatic venous sampling, the clinical team proposed the possibility that multiple, small insulinomas could be present throughout the body of the pancreas. With the patient remaining hypoglycemic, and with few other clinical options, the endocrine surgery team decided to perform a subtotal pancreatectomy and splenectomy with intraoperative ultrasound and frozen section analysis. It was believed that this therapy might help control the patient’s blood sugar levels.
Intra-operative ultrasound revealed a 1cm hypoechogenic lesion within the tail of the pancreas, but no definite mass lesions were appreciated. The pancreatectomy specimen was sent for frozen section diagnosis, and exhaustive sectioning revealed no gross evidence of a lesion. Five representative sections were histologically assessed, and all showed islet hyperplasia of variable size, with no evidence of any discrete lesion. A subtotal pancreatectomy was completed, leaving the uncinate process and a small portion of the pancreatic head in-situ. Within thirty minutes of the procedure, the patient’s blood glucose began to rise, and she was able to be weaned from 20% dextrose in water. In the immediate post-operative period, she remained stable and did not require insulin or glucose administration.
A subtotal pancreatectomy specimen was received for pathologic evaluation which included approximately 75% of the pancreas (head, body and tail measuring 8.0x3.5x1.0 cm) (Figure 1). At the time of frozen section diagnosis the specimen was serially sectioned throughout. No discrete lesions were identified during detailed examination. As previously mentioned, frozen section analysis from five representative areas showed variable-size islet hyperplasia, but no mass lesion was appreciated.
Figure 1. Gross resection specimen. a. Intact
pancreatectomy specimen. b. Sections of pancreas
showing no evidence of a discrete mass or other gross
abnormality.
Extensive additional tissue was taken for permanent section. Histologically, the pancreatic parenchyma showed alteration of the endocrine component in the form of hyperplasia of the islets. Small nests of endocrine cells were present diffusely throughout the pancreatic parenchyma and interlobular septae. Variation in the size and number of islets was striking, and many showed irregular outlines (Figure 2). Occasional islet cells exhibited large hyperchromatic nuclei. There was budding of ducts, forming the ductular-insular complexes characteristic of nesidioblastosis (Figure 2a). The distal pancreas was more severely involved by this process, although the margin of resection was also affected. A separate small focus of chronic pancreatitis was present in one histologic section. Tumor was not identified within the specimen, and the remaining exocrine pancreatic tissue was without abnormalities.
Figure 2. Histologic sections of pancreas. a. Irregular
hyperplastic islets in a haphazard distribution.
Expansion of endocrine cells from a ductal structure
forming a ductulo-insular complex (arrow) (H&E,
20x). b. Irregular hyperplastic islets (arrows) in close
approximation with pancreatic exocrine ductal
structures (H&E, 20x).
Immunohistochemical stains revealed that the islets were composed predominantly of insulin-containing cells, admixed with occasional glucagon, somatostatin, and pancreatic polypeptide-containing cells (Figure 3). There was no specific distribution of these cells within the islets. Considering all gross, histologic, and immunohistochemical findings, a diagnosis of nesidioblastosis was made.
Figure 3. Immunohistochemical stains of pancreas. a.
Chromogranin immunostain demonstrating endocrine
cells within irregular islets (chromogranin, 10x). b.
Insulin immunostain demonstrating predominance of
insulin-secreting cells within the islets (insulin, 20x).
The patient was discharged on post-operative day eight without complications, although she later developed a small left pleural effusion. Over three years later, the patient continues to be followed by the department of endocrinology. She has remained clinically stable, with no significant long-term morbidity post-operatively. She is mildly hypoglycemic at baseline, with glucose levels between 60-80 mg/dL, but is able to manage this through dietary and lifestyle modifications without the use of any medication. One of her offspring has begun to experience a similar constellation of symptoms, and the utility of familial genetic testing is being considered.
DISCUSSION
While the precise incidence of adult nesidioblastosis is unknown, it is clearly a rare entity, with roughly 70 cases described in the literature over three-quarters of a century [5, 6]. Some authors have estimated that 10% of hypoglycemic patients from all age groups with hyperinsulinemia may have nesidioblastosis [6]. However, others have estimated this percentage to be only 0.5-5.0%, particularly in the adult population [7, 8].
The term “nesidioblast” (islet builder) was coined by Laidlaw in 1938, who selected the Greek word for islet, nesidion, and coupled it with blastos to designate the cells that differentiate from ductal epithelium and bud from ducts to form new islets [1, 6]. Laidlaw proposed disregulation of this process to be a possible cause of hypoglycemia, especially in infants [1, 9, 10]. Later, Yakovac et al. wrote that, “nesidioblastosis consists of insulinproducing cells, either isolated, or in small groups of 2-6 cells, dispersed throughout the parenchyma, often in relation to the epithelium of exocrine ducts” [10, 11, 12]. These morphological changes, with distortion of the pancreatic lobular architecture due to endocrine cell proliferation, had been previously described by some authors under the name of nesidiodysplasia [8, 11]. Adult nesidioblastosis may be focal or diffuse, and in some cases chains of endocrine cells may be seen located along the pancreatic ducts, while real islets are not to be found [7]. This has led some authors to consider these lesions a result of inhibited islet differentiation during fetal life [11].
The usual presentation of this condition consists of symptoms related to hypoglycemia, such as dizziness, diaphoresis, tremor, and seizures. Our patient demonstrated repeated bouts of hypoglycemia- induced symptomatology, ultimately culminating in tonic-clonic seizures. Laboratory studies in patients with nesidioblastosis show hypoglycemia and increased insulin and pro-insulin levels in the blood. As outlined in the case report section, our patient’s laboratory studies clearly showed persistent hypoglycemia, with commensurately increased serum insulin and pro-insulin levels. Most patients who present with hyperinsulinemic hypoglycemia outside of the setting of exogenous insulin administration suffer from either insulinoma or nesidioblastosis, whether in its diffuse or focal form [13].
Nesidioblastosis cannot be reliably differentiated from insulinoma by clinical signs and symptoms, and it can be quite difficult to diagnose prior to surgery [9]. The clinical presentation of nesidioblastosis mimics that of insulinoma, and no clear-cut criteria exist to distinguish the various entities. This difficulty makes it mandatory to exclude an insulinoma by all morphologic and functional means before the diagnosis of diffuse nesidioblastosis is established in an adult. After hyperinsulinemia hypoglycemia has been demonstrated by laboratory studies, the presence or absence of a discrete mass lesion on imaging is paramount to directing the differential diagnosis. However, the possibility of so-called occult insulinoma remains in the picture [14]. The use of selective arterial calcium stimulation with hepatic venous sampling can be beneficial in demonstrating hyperactive beta-cell activity [3]. While this methodology may show increased stimulatory function in both insulinomas and nesidioblastosis, it can prioritize the localization and guide resection of pancreatic regions most affected by the process [14, 15]. The technique proved useful in our case, as the appropriate surgical approach was defined pre-operatively, and the patient’s glycemic control was rapidly achieved post-operatively.
Insulinomas are generally discrete mass lesions and typically display a trabecular morphology on microscopic examination. However, a mixed architectural pattern is not uncommon, and many tumors show solid or glandular areas. Most insulinomas have uniform nuclei, with a fine chromatin pattern and only small nucleoli. Nesidioblastosis, in contrast, is histologically characterized by the budding of islet cells from the pancreatic duct epithelium, together with increase in size, shape, and number of the islet cells. Additionally, islet cells are frequently found in unusual locations, such as within the interlobular spaces. As for “islet cell hyperplasia”, some authors have suggested that this terminology should be used to describe a diffuse proliferation of endocrine cells that may express itself with different morphologic patterns, varying from case to case [16]. However, Dahms et al. have defined islet cell hyperplasia as an increased confluent accumulation of abnormally large islets in the center of the lobules, leaving only narrow rims of acinar tissue at some sites [17]. In tissue diagnostic of islet cell hyperplasia, ductular-insular complexes are not to be found, as these are characteristic of nesidioblastosis [17, 18]. Our case did show the typical ductular-insular complexes characteristic of nesidioblastosis, rather than simple islet cell hyperplasia. However, it must be remembered that nesidioblastosis can occur admixed with islet cell hyperplasia. In fact, Kim et al. have reported that only 30 cases of islet cell hyperplasia have been published, and of those only 3 cases consisted solely of hyperplasia without nesidioblastosis [18].
Subtotal (75-90%) pancreatectomy is considered the treatment of choice for nesidioblastosis [13, 16]. Post-surgery, recurrent hypoglycemia or diabetes mellitus are frequent complications. Our patient underwent a subtotal (75%) pancreatectomy and, while doing well in general, has experienced mild persistent hypoglycemia post-operatively.
In conclusion adult nesidioblastosis is a very rare entity which can be difficult to diagnose pre-operatively, as well as at the time of pathologic examination. The clinician must be astute, and the pathologist should be quite certain as to the histologic features of this entity, in order to avoid potential misdiagnosis with other similar entities. The exclusion of insulinoma can be a complex process, and functional studies such as selective arterial calcium stimulation with hepatic venous sampling can be useful in diagnosis and goal-directed surgical intervention.
References
- Laidlaw GF. Nesidioblastoma: the islet tumor of the pancreas. Am J Pathol 1938; 14:125-34.
- Won JG, Tseng HS, Yang AH, Tang KT, Jap TS, Kwok CF, et al. Intra-arterial calcium stimulation test for detection of insulinomas: detection rate, responses of pancreatic peptides, and its relationship to differentiation of tumor cells. Metabolism 2003; 52:1320-9. [PMID 14564685]
- Wiesli P, Brändle M, Schmid C, Krähenbühl L, Furrer J, Keller U, et al. Selective arterial calcium stimulation and hepatic venous sampling in the evaluation of hyperinsulinemic hypoglycemia: potential and limitations. J VascIntervRadiol 2004; 15:1251-6. [PMID 15525744]
- Hussain K, Blankenstein O, De Lonlay P, Christesen HT. Hyperinsulinaemichypoglycaemia: biochemical basis and the importance of maintaining normoglycaemia during management. Arch Dis Child 2007; 92:568-70 [PMID 17588969]
- Jabri AL, Bayard C. Nesidioblastosis associated with hyperinsulinemic hypoglycemia in adults: review of the literature. Eur J Intern Med 2004; 15:407-10. [PMID 15581742]
- Fong TL, Warner NE, Kumar D. Pancreatic nesidioblastosis in adults. Diabetes Care 1989; 12:108- 14. [PMID 2649323]
- Walmsley D, Matheson NA, Ewen S, Himsworth RL, Bevan JS. Nesidioblastosis in an elderly patient. Diabet Med 1995; 12:542-5. [PMID 7648830]
- Gould VE, Chejfec G, Shah K, Paloyan E, Lawrence AM. Adult nesidiodysplasia. Semin Diagn Pathol 1984; 1:43-53. [PMID 6400629]
- Chen YL, Chu JS, Chang TC, Tai TY. Nesidiodysplasia: an unusual cause of hyperinsulinemic hypoglycemia in adults. J Formos Med Assoc 1993; 92:1099-103. [PMID 7911361]
- Yakovac WC, Baker L, Hummeler K. Beta cell nesidioblastosis in idiopathic hypoglycemia of infancy. J Pediatr 1971; 79:226-31. [PMID 4104455]
- Garcia JP, França T, Pedroso C, Cardoso C, Cid MO. Nesidioblastosis in the adult surgical management. HPB Surg 1997; 10:201-9. [PMID 9184873]
- Goudswaard WB, Houthoff HJ, Koudstaal J, Zwierstra RP. Nesidioblastosis and endocrine hyperplasia of the pancreas: a secondary phenomenon. Hum Pathol 1986; 17:46-54. [PMID 2867969]
- Albers N, Löhr M, Bogner U, Loy V, Klöppel G. Nesidioblastosis of the pancreas in an adult with persistent hyperinsulinemic hypoglycemia. Am J Clin Pathol 1989; 91:336-40. [PMID 2646913]
- Tseng LM, Chen JY, Won JG, Tseng HS, Yang AH, Wang SE, Lee CH. The role of intra-arterial calcium stimulation test with hepatic venous sampling (IACS) in the management of occult insulinomas. Ann Surg Oncol 2007; 14:2121-7. [PMID 17431724]
- Draman MS, Ahern T, Smith T, O’Shea D. Selective intra-arterial calcium stimulation with hepatic venous sampling in investigation of hyperinsulinemic hypoglycemia. Ir J Med Sci 2006; 175(Suppl. 2):42.
- Weinstock G, Margulies P, Kahn E, Susin M, Abrams G. Islet cell hyperplasia: an unusual cause of hypoglycemia in an adult. Metabolism 1986; 35:110-7. [PMID 2868380]
- Dahms BB, Landing BH, Blaskovics M, Roe TF. Nesidioblastosis and other islet cell abnormalities in hyperinsulinemic hypoglycemia of childhood. Hum Pathol 1980; 11:641-9. [PMID 7005072]
- Kim YW, Park YK, Park JH, Lee SM, Lee J, Ko SW, Yang MH. Islet cell hyperplasia of the pancreas presenting as hyperinsulinemic hypoglycemia in an adult. Yonsei Med J 2000; 41:426-9. [PMID 10957903]




