Keywords
Cancer; Ca2+ channels; Ca2+ signaling; Cyclic adenosine monophosphate; cAMP signaling
Introduction
Cell signaling is part of a communication process that governs basic activity of cell, and the ability of cell to respond to the microenvironment. This mechanism is fundamental to the homeostasis, tissue repair and control of malignance [1]. Errors in signaling interaction, and cellular information process between cells, are responsible for different pathologies, such as cancer. The development of cancer cells is associated with the mutation of four distinct groups of genes: the protooncogenes growth promoters [2]; tumor suppressor genes [3]; genes that regulate genetically programmed cell death (apoptosis) and genes involved in DNA repair [4]. The abnormal cell division causes cancer, also called as carcinogenesis, and may be associated with exposure to chemicals, radiation [5] or microbial agents, especially viruses [6].
Carcinogenesis is a multi-step process resulting from the accumulation of multiple mutations that accumulate independently in different cell types, generating subclones with different characteristics. These characteristics make the tumors have capacity for invasion and metastasis, rapid growth speed, hormone response and resistance to antineoplastic drugs [7,8]. Numerous normal biochemical mechanisms may be altered, leading to the emergence of these distinct characteristics of cancer cells. Among these several altered biochemical characteristics, the change in the behavior of influx, and efflux, of intracellular Ca2+, and the signaling mediated by cyclic nucleotides i.e., cAMP can be verified. Since intracellular signaling measured by calcium and cyclic nucleotides is a canonical event, changes in this signaling pathway are crucial for the survival and growth of cancer cells [9]. In this way, the knowledge of cancer physiology is crucial to the development of new strategies to control the growth, dissemination and metastasis. In this article, the involvement of Ca2+ channels, and cyclical nucleotides like cAMP in cancer development and progression, and the use of new pharmacological strategies with potential capacity of control the cancer growth, and progression are discussed.
Cyclical nucleotides in cancer cells
The nucleotides are composed by a nitrogenous base, a pentose and one or more phosphate groups, and participate of numerous intracellular biochemical processes. They act as precursors of deoxyribonucleic acid (DNA) and ribonucleic acid (RNA), energy source (adenosine triphosphate and guanosine triphosphate), coenzymes (flavin adenine dinucleotide, nicotinamide adenine dinucleotide and coenzyme A) and physiological regulators (cyclic adenosine monophosphate and cyclic guanosine monophosphate) [10]. The cyclic adenosine monophosphate (cAMP) is a second messenger that acts as intracellular signal transduction leading to a cAMP-dependent pathway. The cAMP is synthesized from ATP by the adenylyl cyclase located on the inner side of the plasma membrane.
Adenylyl cyclase is activated by signaling molecules through the activation of receptors with the G-protein stimulatory (Gs) of adenylyl cyclase, and inhibited by inhibitory G (G) receptor agonists of adenylyl cyclase [11]. When cAMP concentration increases (activation of the adenylate cyclases by the Gs protein, and inhibition of cAMP-degrading phosphodiesterases), cAMP binds to the regulatory subunits, which leads to the release of the catalytic subunits. The free catalytic subunits catalyze the transfer of terminal phosphates from ATP [12]. The link between membrane surface of cell and cytoplasm is mediated by a family of enzymes called kinase proteins dependent of cAMP, or protein kinase A (PKA), by transformation of ATP in ADP with phosphorylation of protein substrates responsible by intracellular effects [13]. Mechanisms involving the control of cAMP over PKA can be divided into: direct protein phosphorylation and protein synthesis. In direct phosphorylation, PKA both increases and decreases the activity of a protein; and in protein synthesis PKA first activates the cAMP response element-binding protein (CREB), a cellular transcription factor, which binds to the cAMP response element, altering transcription and protein synthesis [14]. In the negative regulation of PKA, one of the substrates activated by kinase is a phosphodiesterase, which converts cAMP to AMP, reducing the amount of cAMP that can activate PKA. The catalytic function of PKA can be combined with Akinase anchoring proteins (AKAP). AKAP are signal-organizing molecules that compartmentalize various enzymes that are regulated by second messengers. PKA binding with AKAP, and a phosphodiesterase, form a complex that hydrolyzes cAMP. Considering the phosphodiesterase contributes to the low concentration of cAMP in cells, PKA is responsible for the activation of phosphodiesterase, to lower the concentration of cAMP [11]. The cyclic nucleotides, cyclic adenosine monophosphate (cAMP) and cyclic guanosine monophosphate (cGMP), are important intracellular signal transduction molecules, acting as second messengers through an extracellular signal. Both cAMP and cGMP signaling have positive or negative effects on growth and survival, depending on the type cell. The cAMP can regulate a variety of cellular functions: metabolism of ion channel activation, cell growth and differentiation, gene expression and apoptosis [15]. The cAMP pathway acts with other intracellular signaling pathways such as those mediated by Ca2+ [16], and Jak/STAT [17]. The cAMP interacts with Ras-mediated MAP kinase, modulating cell growth [18] when binding to cAMP-dependent protein kinases (PKA) [19]. When activated, PKA phosphorylates macromolecular complexes responsible for the destruction of mitotic cyclins, and separation of sister chromatids in the anaphase-metaphase transition [20]. The involvement of cAMP, and the activation of PKA, has been associated with different types of cancer [21], where oncogenic activity of cAMP is due to the activation of PKA, and downstream effectors (exchange protein directly activated by cAMP (Epac) and CREB) [22].
The PKA-mediated cascade is required for the functional regulation of D-type cyclins, so defects in the cAMP/PKA pathway can induce tumors in cell lines [23], which can be reversed by modifying the PKA subunit type that is expressed by the cell. The circuity formed by PKA, and cAMP, can influence the growth of colorectal cancer cell by decreasing cAMP intracellular levels [24]. Any tumors present a predominant of determined forms of PKA, such as glioblastoma, with predomin of PKA type II [25]. In the same way, the increase of cAMP levels can diminish the tumor growth [26]. Other function, in which PKA may be dysregulated in cancer, is the cell migration that involves cytoskeleton remodeling [27].
Numerous mutations lead to the formation of oncogenes that encode different protein kinases. Changes in the activity of protein kinases alter numerous signaling pathways, such as those involved in the cytosolic concentration of Ca2+. Intracellular signals mediated by abnormal cytosolic Ca2+ concentrations are important in maintenance, growth, inavasion and metastasis by cancer cells.
Ca2+ signaling and channels in cancer cells
The Ca2+ acts as an important intracellular messenger because it is a bivalent molecule that has strong and specific binding to it receptor, and has an atomic radius that gives it ideal geometry for protein binding [28]. Usually, Ca2+ is stored in specific organelles, such as endoplasmic reticulum and mitochondria [9]. Indeed, intracellular Ca2+ homeostasis is regulated by numerous channels and transporters of Ca2+, for example by the receptor of inositol-1,4,5-triphosphate (IP3R) and Ca2+-ATPase pump [for example plasma membrane Ca2+- ATPase (PMCA), ER/SR Ca2+-ATPase (SERCA), and golgi vesicles secretory pathway Ca2+-ATPase (SPCA)]. In addition, the Ca2+ influx across plasma membrane occurs through voltageactivated Ca2+ channels (VACCs, also known as Cav family) and transient receptor potential channels (TRPs). Intracellular Ca2+ homeostasis is also regulated by the Ca2+-induced Ca2+ release (CICR) mechanism, Na+/Ca2+ exchanger (NCX) and mitochondrial Ca2+ uniporter (MCU) [29].
The release of Ca2+ from the endoplasmic reticulum to the cytoplasm is performed through classical signalling pathways, activated by specific agonists and receptors, located in the surface of plasma membrane, for example by activating phospholipase C, it hydrolyzes phosphatidylinositol 4,5- bisphosphate (PIP2) of plasma membrane, so producing inositol-1,4,5-triphosphate (IP3). The diffusion of IP3 into the cell releases intracellular Ca2+ of their stocks by the activation of specific receptors (IP3R), which are localized in the cytoplasmic side of endoplasmic reticulum membrane [30]. The increase of expression, or activity, of Ca2+ channels in the plasma membrane leads to increase of Ca2+ influx, promoting Ca2+-dependent cell proliferation and differentiation [31]. These mechanisms of influx, and efflux, of intracellular Ca2+ are dependent on Ca2+ transporters located mostly in the plasma membrane. Several Ca2+ channels, like Ca2+-dependent voltage channels, are involved in the Ca2+ influx. However, these channels require depolarization of the plasma membrane, being more common on the surface of excitable cells, as cardiomyocytes [32]. Some of these Ca2+ channels, are members of the Cav3 subfamily activated by low voltage, are expressed on the surface of different cancerous cells [33], and Ca2+ entry in non-excitable cells mostly occurs through nonvoltage gated channels.
The non-voltage gated channels of Ca2+ associated with different types of cancer cells include: ligand-gated channels; receptor-operated channels (ROC) or secondary messengeroperated channels linked to GPCR activation [SMOC: Orai family and members of TRP (Transient Receptor Potential) superfamily of channels]; store-operated channels (SOCE: Orai family and members of TRPC (TRP Canonical) subfamily of channels); and stretch-operated channels (members of TRP superfamily of channels); plasma membrane Ca2+-ATPase (PMCA); and Na+/Ca2+ exchanger [34].
Cellular proliferation depends on the cell cycle, which is dependent on Ca2+. Cell proliferation, and cell division, depend on extracellular Ca2+, and the increase in intracellular Ca2+ is involved in cell cycle progression, and proliferation [35]. The Ca2+ is required at the beginning of the G1 phase of the cell cycle, where activation of transcription factors like activator protein 1 (AP1), a transcription factor that regulates gene expression, and cellular processes differentiation, proliferation, and apoptosis; cAMP-responsive element binding protein (CREB) and the nuclear factor of activated Tcell (NFAT) [36].
The Ca2+ plays a key role in the expression of cell cycle regulators like the D-type cyclins, required for the activation of cyclin-dependent kinase 4 complexes, responsible of phosphorylation and inactivation of retinoblastoma gene, involved in the entry into S phase of cell cycle. The start of G1/S phase is dependent of Ca2+ calmodulin (CaM), and CaMkinase II (CaMK) [37]. Calcineurin, a Ca2+-dependent phosphatase, plays a major role in progression of G1 and S phases, regulating cyclins A, D1 and E [37,38] and active NFAT, favoring the cell proliferation, through the activation of Ca2+ channels. The IP3Rs are the major channels of intracellular Ca2+ release in non-excitable cells, being activated in different types of cancer, such as gastric and colorectal cancer [39,40]. Making part of the Ca2+-ATPases family, SERCA presents an altered expression in diferents cancers cells such as colon, gastric, lung, myeloid leukaemia and choroid plexus [41]. Altered expression of SPCA isoforms are expressed in breast, colon and prostate cancer [42], and altered expression of PMCA isoforms are expressed in breast cancer cells [43].
The non-voltage gated channels of Ca2+, Orai and stromal interaction molecule 1 (STIM1), a Ca2+ sensor in the endoplasmic reticulum, present higher expression in glioblastoma [44], pancreatic adenocarcinoma [45], prostate cancer [46] and hepatocellular carcinoma [47]. The MCU is overexpressed in breast cancer cells [48]. Changes in expression of TRP channels, like TRPV1, TRPV2, TRPV6, TRPM8, TRPM2, TRPC6 [34], L-type calcium channel [49], and T-type Ca2+ channels [50,51] were observed in prostate cancer cells. Also, the expression of TRP channels TRPC1, TRPC3, TRPC6, TRPM7, TRPM8, and TRPV6 is altered in breast cancer [52], thyroid, colon and ovary cancer, with emphasis of TRPV6 [53,54]. In lung cancer cells the expression of TRPC1, TRPC3, TRPC4, TRPC6, TRPM7, and TRPM8 is altered [55]. During the process of metastasis, Ca2+ participles of invasion of health tissues by cancer cells, with involvement of voltage independent Ca2+ channels [56-58] in breast [59,60] and lung cancer cell [61].
Potential use of modulators of cyclical nucleotides or inhibitors of Ca2+ channels
The growing understanding of cancer biology has led to the development of new drugs for the treatment of cancer. However, a total beneficial effect of these agents has not yet been verified by the presence of intrinsic cancer cell resistance, the result of compensatory signaling pathways, or the development of acquired resistance through the evolution of cell clones by selective treatment pressures.
Recognizing the toxicity induced by the treatment and the inability to use high effective pharmacological doses in the treatment of cancer, we are exploring the combination of Ca2+ channel blockers and/or enhancer agents of cAMP, associated with chemotherapy, radiotherapy or immunotherapy.
Variations in expression of Ca2+ channels in cell cancer suggest that a decrease in Ca2+ channel expression, or Ca2+ influx, will lead to cell cycle arrest, inhibiting the process of invasion, metastasis, and recurrence of cancer. We also believe that increasing the cytosolic concentration of cAMP produced by the drug combination could simultaneously generate activation of the RAS (antitumor) mediated signaling pathway, and inhibition of the PKA (pro-tumor) pathway, favoring the host.
For example, new treatments of cancer involving the use of monoclonal antibodies against programmed death 1 (PD-1) receptor, and its PD-L1 ligand [62], presented promising results. PD-L1 is expressed in different types of cancer cells, such melanoma, lung, breast, ovaries, pancreas, esophagus, bladder and haematological tumors [63]. However, in spite of the positive results observed with the use of monoclonal antibodies against PD-L1/PD-1, recent studies have revealed an aggressive growth of tumors in a small portion of patients [64,65]. This process of tumor progression after immunotherapy has been described as being associated with amplification of MDM2/MDM4 genes [65]. The MDM2/MDM4 genes inhibit the p53 tumor suppressor gene [66]. Normally p53 is activated in response to DNA damage, or oncogene activation, which in turn starts mechanisms of apoptosis, cellcycle arrest or modulation of autophagy.
Monoclonal antibodies against PD-1 can induce the increase synthesis of interferon gamma (IFN-γ) by T lymphocytes [67], which in turn activates JAK-STAT signaling [68] resulting in increase of interferon regulatory fator-8 (IRF8) expression [69]. Finally, the IRF8 binds to the MDM2 promoter inducing MDM2 higher expression [70] shown in Figure 1A.
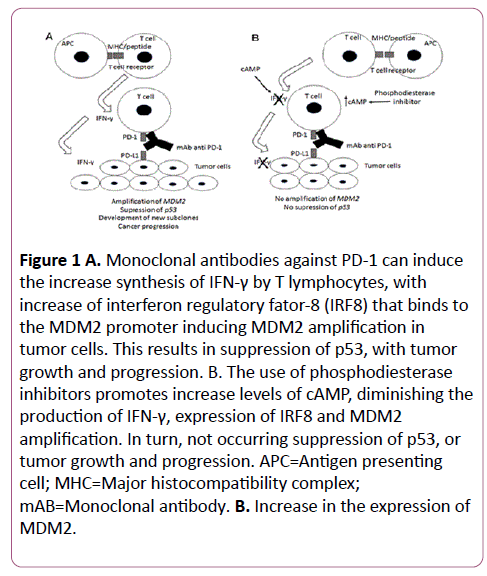
Figure 1:A. Monoclonal antibodies against PD-1 can induce the increase synthesis of IFN-γ by T lymphocytes, with increase of interferon regulatory fator-8 (IRF8) that binds to the MDM2 promoter inducing MDM2 amplification in tumor cells. This results in suppression of p53, with tumor growth and progression. B. The use of phosphodiesterase inhibitors promotes increase levels of cAMP, diminishing the production of IFN-γ, expression of IRF8 and MDM2 amplification. In turn, not occurring suppression of p53, or tumor growth and progression. APC=Antigen presenting cell; MHC=Major histocompatibility complex; mAB=Monoclonal antibody. B. Increase in the expression of MDM2.
Because the elevation of intracellular cAMP creates an oxidative environment that oxidizes and inactivates p56(lck) in lymphocytes by an H2O2 dependent, PKA-independent mechanism, and inhibits the production of IFN-γ by nitric oxide, PKA-dependent mechanism [71], the use of phosphodiesterase inhibitors can promote the increase levels of cAMP [72], hindering the production of IFN-γ, which would make difficult to increase the expression of IRF8, and increase in the expression of MDM2 shown in Figure 1B.
Thus, the combination of a phosphodiesterase inhibitor with anti-PD-1 monoclonal antibodies could prevent the emerging of new more aggressive tumor subclones.
This new pharmacological strategy could be extended not only for the use of modulators of cAMP, but also for inhibitors of Ca2+ channels. For example, it was described that the use of inhibitor of Ca2+-dependent K+ channels (TRAM-34) is able to block the growth of hepatocellular carcinoma [73]. Thus, the use of TRAM-34 may be associated with hepatic intra-arterial chemotherapy, allowing a minor concentration of the chemotherapeutic floxuridine [74] in the liver with a lower systemic toxic effect to the treatment of hepatic adenocarcinoma, primary or metastatic. Because the relevance of Orai1 and TRP channels in tumor neovascularization [75], blockers of these channels can diminish the adverses effects of treatment with ramucirumab, a monoclonal antibodies against vascular endotelial growth factor receptor 2 (VEGFR2), with anti-angiogenic effect used to the treatment of advanced gastric, gastro-oesophageal junction adenocarcinoma and non-small cell lung cancer (NSCLC), with the possibility of decreased toxicity, and adverses effects like neutropenia, febrile neutropenia and hypertension [76].
Conclusion
Thus, the use of modifiers of cAMP production may decrease the chance of developing intrinsic anti-tumor resistance, and the use of Ca2+ channel blockers may modify tumor growth, and also by reducing the adverse effects of chemotherapy, or immunotherapy.
References
- Carling D (2017) AMPK signalling in health and disease. CurrOpin Cell Biol 45: 31-37.
- Hnisz D, Weintraub AS, Day DS, Valton AL, Bak RO, et al. (2016) Activation of proto-oncogenes by disruption of chromosome neighborhoods. Science 351: 1454-1458.
- Eshghifar N, Farrokhi N, Naji T, Zali M (2017) Tumor suppressor genes in familial adenomatous polyposis. GastroentHepatol Bed Bench 10: 3-13.
- Uchida C (2016) Roles of pRB in the regulation of nucleosome and chromatin structures. Biomed Res Int.
- Zegarska B, Pietkun K, Zegarski W, Bolibok P, Wiśniewski M, et al. (2017) Air pollution, uv irradiation and skin carcinogenesis: what we know, where we stand and what is likely to happen in the future? Postepy Dermatol Alergol 34: 6-14.
- Yajid AI, Zakariah MA, Mat Zin AA, Othman NH (2017) Potential role of E4 protein in human papillomavirus screening: a Review. Asian Pac J Cancer Prev 18: 315-319.
- Ellsworth DL, Blackburn HL, Shriver CD, Rabizadeh S, Soon-Shiong P, et al. (2017) Single-cell sequencing and tumorigenesis: improved understanding of tumor evolution and metastasis. Clin Transl Med 6: 15.
- Kachalaki S, Ebrahimi M, Khosroshahi ML, Mohammadinejad S, Baradaran B (2016) Cancer chemoresistance; biochemical and molecular aspects: a brief overview. Eur J Pharm Sci 89: 20-30.
- Errante PR, Neto AC, Bergantin LB (2017) Insights for the inhibition of cancer progression: Revisiting Ca2+ and cAMP signalling pathways. Adv Cancer Prev 2: e103.
- Yan K, Gao LN, Cui YL, Zhang Y, Zhou X (2016) The cyclic AMP signaling pathway: exploring targets for successful drug discovery. Mol Med Rep 13: 3715-3723.
- Sharma RK, Duda T, Makino CL (2016) Integrative signaling networks of membrane guanylate cyclases: biochemistry and physiology. Front Mol Neurosci 9: 83.
- Berisha F, Nikolaev VO (2017) Cyclic nucleotide imaging and cardiovascular disease. PharmacolTherpii: S0163-7258(17)30052-9.
- Palorini R, Votta G, Pirola Y, De Vitto H, De Palma S, et al. (2016) Protein kinase A activation promotes cancer cell resistance to glucose starvation. Anoikis PLoS Genet 12: e1005931.
- Yan K, Gao LN, Cui YL, Zhang Y, Zhou X (2016) The cyclic AMP signaling pathway: exploring targets for successful drug discovery. Mol Med Rep 13: 3715-3723.
- Chin KV, Yang WL, Ravatn R, Kita T, Reitman E, et al. (2002) Reinventing the wheel of cyclic AMP: Novel mechanisms of cAMP signaling. Ann NY Acad Sci 968: 49-64.
- Rogue PJ, Humbert JP, Meyer A, Freyermuth S, Krady MM, et al. (1998) cAMP-dependent protein kinase phosphorylates and activates nuclear Ca2+-ATPase. Proc Natl Acad Sci USA 95: 9178-9183.
- David M, Petricoin E, Larner AC (1996) Activation of protein kinase A inhibits interferon induction of the Jak/Stat pathway in U266 cells. J Biol Chem 271: 4585-4588.
- Cook SJ, McCormick F (1993) Inhibition by cAMP of Ras-dependent activation of Raf. Science 262: 1069-1072.
- Stork PJ, Schmitt JM (2002) Crosstalk between cAMP and MAP kinase signaling in the regulation of cell proliferation. Trends Cell Biol 12: 258-266.
- Ferrari S (2006) Protein kinases controlling the onset of mitosis. Cell Mol Life Sci 63: 781-795.
- Caretta A, Mucignat-Caretta C (2011) Protein kinase A in cancer. Cancers 3: 913-926.
- Borland G, Smith BO, Yarwood SJ (2009) EPAC proteins transduce diverse cellular actions of cAMP. Br J Pharmacol 158: 70-86.
- Prasad KN, Cole WC, Yan XD, Nahreini P, Kumar B, et al. (2003) Defects in cAMP-pathway may initiate carcinogenesis in dividing nerve cells: A review. Apoptosis 8: 579-586.
- Cho-Chung YS, Nesterova M, Becker KG, Srivastava R, Park YG, et al. (2002) Dissecting the circuitry of protein kinase A and cAMP signaling in cancer genesis: antisense, microarray, gene overexpression, and transcription factor decoy. Ann NY Acad Sci 968: 22-36.
- Frattola L, Canal N, Ferrarese C, Tonini C, Tonon G, et al. (1983) Multiple forms of protein kinase from normal human brain and glioblastoma. Cancer Res 43: 1321-1324.
- Hanson AJ, Nahreini P, Andreatta C, Yan XD, Prasad KN (2005) Role of the adenosine 3’,5’-cyclic monophosphate (cAMP) in enhancing the efficacy of siRNA-mediated gene silencing in neuroblastoma cells. Oncogene 24: 4149-4154.
- Howe AK (2004) Regulation of actin-based cell migration by cAMP/PKA. BiochimBiophys Acta 1692: 159-174.
- Decrock E, Hoorelbeke D, Ramadan R, Delvaeye T, De Bock M et al. (2017) Calcium, oxidative stress and connexin channels, a harmonious orchestra directing the response to radiotherapy treatment? BiochimBiophys Acta pii: S0167-4889(17)30032-0.
- Cui C, Merritt R, Fu L, Pan Z (2017) Targeting calcium signaling in cancer therapy. Acta PharmaceuticaSinica B 7: 3-17.
- Resende RR, Andrade LM, Oliveira AG, Guimarães ES, Guatimosim S, et al. (2013) Nucleoplasmatic calcium signaling and cell proliferation: calcium signaling in the nucleus. Cell Commun Signal. 11: 14.
- Roderick HL, Cook SJ (2008) Ca2+ signaling checkpoints in cancer: remodeling Ca2+ for cancer cell proliferation and survival. Nat Rev Cancer 8: 361-375.
- Song Z, Ko CY, Nivala M, Weiss JN, Qu Z (2015) Calcium-voltage coupling in the enesis of early and delayed after depolarizations in cardiac myocytes. Biophys 108: 1908-1921.
- Prevarskaya N, Skryma R, Shuba Y (2010) Ion channels and the hallmarks of cancer. Trends Mol Med 16: 107-121.
- Deliot N, Constantin B (2015) Plasma membrane calcium channels in cancer: Alterations and consequences for cell proliferation and migration. Biochimica et Biophysica Acta 1848: 2512-2522.
- Prakriya M, Lewis RS (2015) Store-operated calcium channels. Physiol Rev 95: 1383-1436.
- Parkash J, Asotra K (2010) Calcium wave signaling in cancer cells. Life Sci 87: 587-595.
- Kahl CR, Means AR (2003) Regulation of cell cycle progression by calcium/calmodulin dependent pathways. Endocr Rev 24: 719-736.
- Tomono M, Toyoshima K, Ito M, Amano H, Kiss Z (1998) Inhibitors of calcineurin block expression of cyclins A and E induced by fibroblast growth factor in Swiss 3T3 fibroblasts. Arch Biochem Biophys 353: 374-378.
- Sakakura C, Hagiwara A, Fukuda K, Shimomura K, Takagi T, et al. (2003) Possible involvement of inositol 1,4,5-trisphosphate receptor type 3 (IP3R3) in the peritoneal dissemination of gastric cancers. Anticancer Res 23: 3691-3697.
- Shibao K, Fiedler MJ, Nagata J, Minagawa N, Hirata K, et al. (2010) The type III inositol 1,4,5-trisphosphate receptor is associated with aggressiveness of colorectal carcinoma. Cell Calcium 48: 315-323.
- Dang D, Rao R (2016) Calcium-ATPases: gene disorders and dysregulation in cancer. BiochimBiophys Acta 1863: 1344-1350.
- Monteith GR, Davis FM, Roberts-Thomson SJ (2012) Calcium channels and pumps in cancer: changes and consequences. J Biol Chem 287: 31666-31673.
- Lee WJ, Roberts-Thomson SJ, Monteith GR (2005) Plasma membrane calcium-ATPase 2 and 4 in human breast cancer cell lines. Biochem Biophys Res Commun 337: 779-783.
- Motiani RK, Hyzinski-Garcia MC, Zhang X, Henkel MM, Abdullaev IF, et al. (2013) STIM1 and Orai1 mediate CRAC channel activity and are essential for human glioblastoma invasion. Pflugers Arch 465: 1249-1260.
- Kondratska K, Kondratskyi A, Yassine M, et al. (2014) Orai1 and STIM1 mediate SOCE and contribute to apoptotic resistance of pancreatic adenocarcinoma. BiochimBiophys Acta 1843: 2263-2269.
- Dubois C, VandenAbeele F, Lehen'kyi V, et al. (2014) Remodeling of channel-forming ORAI proteins determines naoncogenics witch in prostate cancer. Cancer Cell 26: 19-32.
- Yang N, Tang Y, Wang F, et al. (2013) Blockade of store-operated Ca2+ entry inhibits hepatocarcinoma cell migration and invasion by regulating focal adhesion turnover. Cancer Lett 330: 163-169.
- Curry MC, Peters AA, Kenny PA, et al. (2013) Mitochondrial calcium uniporter silencing potentiates caspase-independent cell death in MDA-MB-231 breast cancer cells. Biochem Biophys Res Commun 434: 695-700.
- Chen R, Zeng X, Zhang R, et al. (2014) Cav1.3 channel alpha1D protein is overexpressed and modulates androgen receptor transactivation in prostate cancers. Urol Oncol 32: 524-536.
- Haverstick DM, Heady TN, Macdonald TL, et al. (2000) Inhibition of human prostate cancer proliferation in vitro and in a mouse model by a compound synthesized to block Ca2+ entry. Cancer Res 60: 1002-1008.
- Mariot P, Vanoverberghe K, Lalevee N, et al. (2002) Overexpression of an alpha 1H (Cav3.2) T-type calcium channel during neuroendocrine differentiation of human prostate cancer cells. J Biol Chem 277: 10824-10833.
- Chen J, Luan Y, Yu R, et al. (2014) Transient receptor potential (TRP) channels, promising potential diagnostic and therapeutic tools for cancer. Biosci Trends 8: 1-10.
- Zhuang L, Peng JB, Tou L, et al. (2002) Calcium-selective ion channel, CaT1, is apically localized in gastrointestinal tract epithelia and is aberrantly expressed in human malignancies. Lab Investig 82: 1755-1764.
- Lehen'kyi V, Raphael M, Prevarskaya N (2012) The role of the TRPV6 channel in cancer. J Physiol 590: 1369-1376.
- Jiang HN, Zeng B, Zhang Y, et al. (2013) Involvement of TRPC channels in lung cancer cell differentiation and the correlation analysis in human non-small cell lung cancer. PLoS One 8: e67637.
- Wei C, Wang X, Chen M, et al. (2010) Flickering calcium microdomains signal turning of migrating cells. Can J Physiol Pharmacol 88: 105-110.
- Wei C, Wang X, Chen M, et al. (2009) Calcium flickers steer cell migration. Nature 457: 901-905.
- Monet M, Lehen'kyi V, Gackiere F, et al. (2010) Role of cationic channel TRPV2 in promoting prostate cancer migration and progression to androgen resistance. Cancer Res 70: 1225-1235.
- Hammadi M, Chopin V, Matifat F, et al. (2012) Human ether a-gogo K(+) channel 1 (hEag1) regulates MDAMB-231 breast cancer cell migration through Orai1-dependent calciumentry. J Cell Physiol 227: 3837-3846.
- Yang S, Zhang JJ, Huang XY (2009) Orai1 and STIM1 are critical for breast tumor cell migration and metastasis. Cancer Cell 15: 124-134.
- Gao H, Chen X, Du X, et al. (2011) EGF enhances the migration of cancer cells by up-regulation of TRPM7. Cell Calcium 50: 559-568.
- Gravelle P, Burroni B, Péricart S, et al. (2017) Mechanisms of PD-1/PD-L1 expression and prognostic relevance in non-Hodgkin lymphoma: a summary of immunohistochemical studies. Oncotarg.
- Zitvogel L, Kroemer G (2012) Targeting PD-1/PDL1 interactions for cancer immunotherapy. Oncoimmunol 1:1223-1225.
- Champiat S, Dercle L, Ammari S, et al. (2017) Hyperprogressive disease is a new pattern of progression in cancer patients treated by anti-PD-1/PD-L1. Clin Cancer 23: 1920-1928.
- Kato S, Goodman AM, Walavalkar V, et al. (2017) Hyper-progressors after immunotherapy: Analysis of genomic alterations associated with accelerated growth rate. Clin Cancer Res 2: 3133.
- Wade M, Li YC, Wahl GM (2013) MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat Rev Cancer 13: 83-96.
- Peng W, Liu C, Xu C, et al. (2012) PD-1 blockade enhances T-cell migration to tumors by elevating IFN-gamma inducible chemokines. Cancer Res 72: 5209-5218.
- Schindler C, Levy DE, Decker T (2007) JAK-STAT signaling: from interferons to cytokines. J Biol Chem 282: 20059-20063.
- Waight JD, Netherby C, Hensen ML, et al. (2013) Myeloid-derived suppressor cell development is regulated by a STAT/IRF-8 axis. J Clin Invest 123: 4464-4478.
- Zhao Y, Yu H, Hu W (2014) The regulation of MDM2 oncogene and its impact on human cancers. Acta BiochimBiophys Sin 46: 180-189.
- Cochrane R, Clark RB, Huang CK, et al. (2001) Differential regulation of T cell receptor-mediated Th1 cell IFN-gamma production and proliferation by divergent cAMP-mediated redox pathways. J Interferon Cytokine Res 21: 797-807.
- Xiao J, Sun Q, Bei Y, et al. (2016) Therapeutic inhibition of phospholipase D1 suppresses hepatocellular carcinoma. Clin Sci 130: 1125-1136.
- Freise C, Ruehl M, Seehofer D, et al. (2013) The inhibitor of Ca2+-dependent K+ channels TRAM-34 blocks growth of hepatocellular carcinoma cells via downregulation of estrogen receptor alpha mRNA and nuclear factor-kappaB. Invest New Drugs 31: 452-457.
- Cercek A, Boucher TM, Gluskin JS, et al. (2016) Response rates of hepatic arterial infusion pump therapy in patients with metastatic colorectal cancer liver metastases refractory to all standard chemotherapies. J Surg Oncol 114: 655-663.
- Moccia F, Dragoni S, Poletto V, et al. (2014) Orai1 and transient receptor potential channels a novel molecular targets to impair tumor neovascularization in renal cell carcinoma and other malignancies. Anticancer Agents Med Chem 14: 296-312.
- Oscar A, Zyanya Lucia ZB, Andres FC, et al. (2017) Ramucirumab in the treatment of non-small cell lung cancer. Expert Opin Drug Saf