Keywords
Functional gastrointestinal disorders; Immunohistochemistry; Intractable diarrhea of infancy; Lactic acidosis; Mitochondrial respiratory chain deficiencies
Introduction
Mitochondrial respiratory chain disorders (MRCDs) are metabolic disorders caused by defects in the genes that encode the enzymes composing the mitochondrial respiratory chain. Their prevalence has been reported as 1 per 5,000 births [1]. MRCDs may affect a variety of organs, and their clinical symptoms are correspondingly diverse [2]. For instance, the symptoms of mitochondrial neurogastrointestinal encephalopathy include dysfunction of gastrointestinal tract movement, peripheral neuropathy, and leukoencephalopathy [3-5]. MRCDs usually become apparent soon after birth; it is rare for manifestations to be delayed until the first year of life. However, herein we present such a case: A 13-month-old girl who was admitted to our hospital with frequent vomiting and later developed intractable diarrhea. Although the diagnosis was difficult and the symptoms were treatment-resistant, we suspected MRCD because of the presence of hyperlactatemia and liver dysfunction. We performed a colonoscopy and immunostaining of mitochondrial complexes in the colonic mucosa, which revealed that MRC complex I was weakly expressed. Accordingly, the patient was diagnosed with MRC complex I deficiency.
Case Report
A 13-month-old girl with an unremarkable medical history developed fever on day 1 and frequent vomiting the day after that. She vomited blood from the onset of the complaints until the 3rd day and was admitted to the community hospital on the 3rd day. As her symptoms were consistent with those of Mallory-Weiss syndrome due to gastroenteritis, she was initially treated by being placed on a total fast with no food or drink. Whole-body computed tomography showed no abnormality. When admitted in the community hospital, her nutritional needs were taken care of through an intravenous infusion. On the 11th day of illness, she was transferred to our hospital for detailed investigation. Her fever lasted for 20 days, which was consistent with a diagnosis of fever from unknown causes.
Her height and weight were 70 cm (-1.3 SD) and 7.8 kg (-1.8 SD), respectively, as measured at the time of admission. Her consciousness was clear. Her respiratory sounds were clear and her breathing unlabored. Her heart sounds were normal, with no heart murmur. Abdominal palpation revealed no hepatosplenomegaly or tenderness. Her skin showed severe atopic dermatitis, with moist eczema ranging from her face to her trunk, hands, and feet.
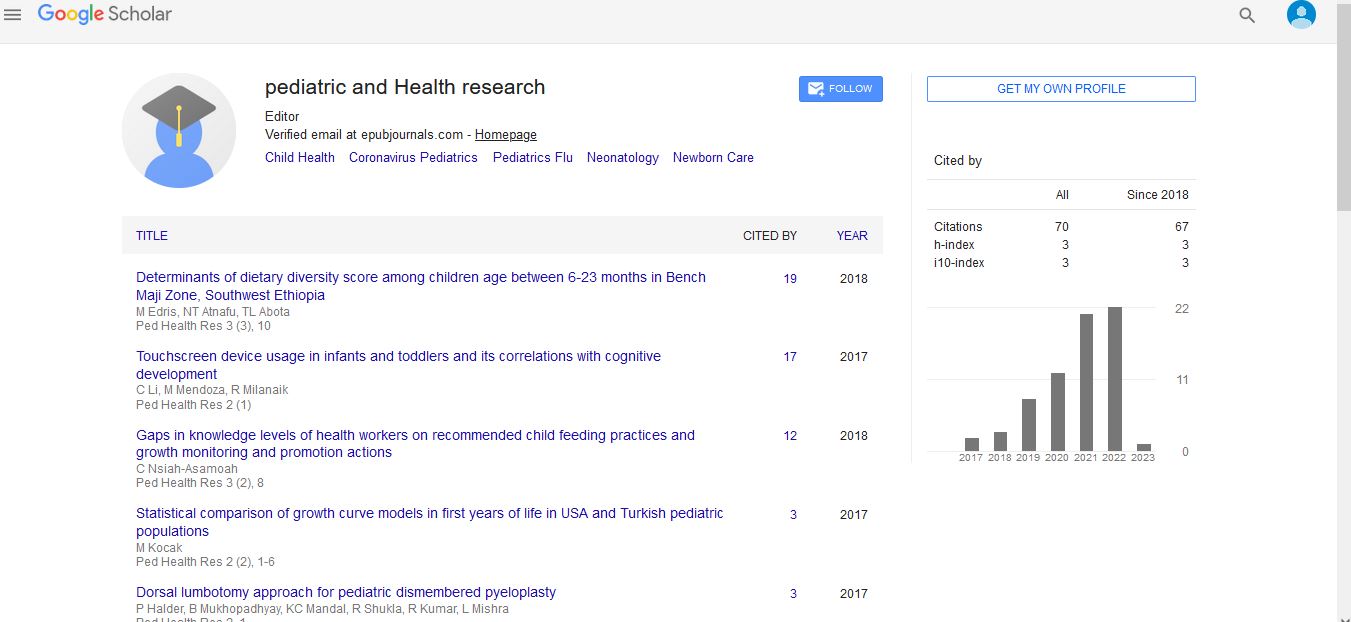
Blood tests revealed a white blood cell count of 5.8 × 103/mm3, 1.6 × 103/mm3 of neutrophils, a C-reactive protein level of 0.01 mg/dL, and a blood lactate level of 24.5 mg/dL (normal range 3 mg/dL to 17 mg/dL). Tandem mass spectrometry for fatty acids and organic acids showed no obvious abnormality. Immunological tests indicated neutrophil antimicrobial activity of 97.9%, a CD4/CD8 ratio of 2.28, and natural killer cell activity of 27%. Serum levels of immunoglobulin G, A and M were 428 mg/dL, 40 mg/dL and 102 mg/dL, respectively. An abdominal radiograph suggested having a large volume of gas but no air-fluid levels. Gastrointestinal contrast radiography suggested a gastric volvulus without evidence of obstruction, stenosis, or a spaceoccupying lesion (Figures 1a and 1b). Esophagogastroduodenoscopy revealed some redness in the esophagus, but there were no obvious sources of bleeding, such as ulcers or erosive lesions, in the stomach and duodenum (Figures 2a and 2b).
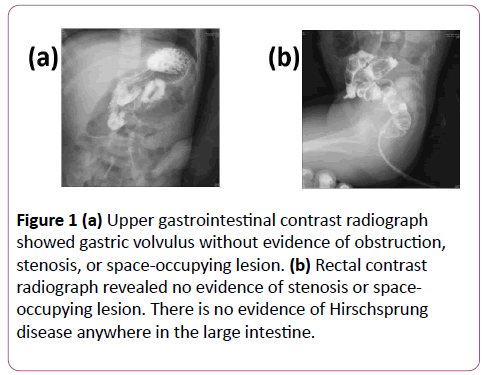
Figure 1: (a) Upper gastrointestinal contrast radiograph showed gastric volvulus without evidence of obstruction, stenosis, or space-occupying lesion. (b) Rectal contrast radiograph revealed no evidence of stenosis or spaceoccupying lesion. There is no evidence of Hirschsprung disease anywhere in the large intestine.
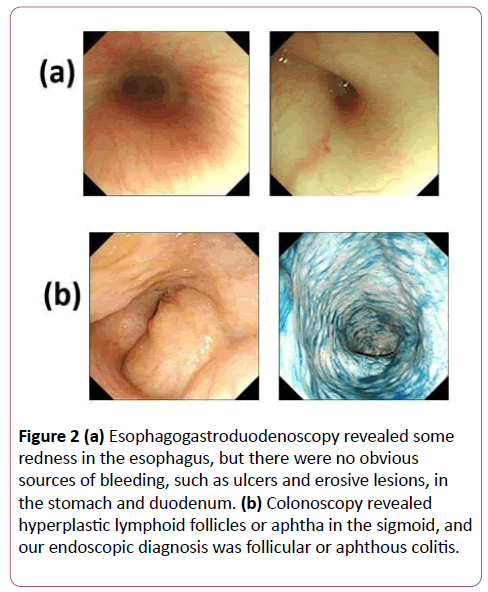
Figure 2: (a) Esophagogastroduodenoscopy revealed some redness in the esophagus, but there were no obvious sources of bleeding, such as ulcers and erosive lesions, in the stomach and duodenum. (b) Colonoscopy revealed hyperplastic lymphoid follicles or aphtha in the sigmoid, and our endoscopic diagnosis was follicular or aphthous colitis.
Food allergies were suspected based on the fact that the patient had eaten baby food just prior to admission to our hospital, and the patient was switched to an elemental diet, Elental P® (Ajinomoto, Tokyo, Japan; lipid 8%) on the 16th day, administered through a duodenal catheter. Although her skin improved gradually, her vomiting did not improve, and she began having frequent diarrhea from the 30th day onward. We considered the possibility of soy allergy and switched to soyfree Elemental Formula (Meiji, Tokyo, Japan; lipid 6%) on the 25th day, but her vomiting and diarrhea did not abate.
To investigate the causes of fever of unknown origin, vomiting, and diarrhea, we performed the following examinations. Cytomegalovirus enteritis was not suspected because cytomegalovirus DNA was not detected in the feces. The results of ocular fundus examination were inconsistent with Behçet's disease. We conducted a colonoscopy with biopsy on the 53rd day, during which endoscopic examination revealed nonspecific lymphoid hyperplasia in the sigmoid colon, indicating follicular or aphthous colitis (Figure 2), and histological examination showed nonspecific inflammation of the colonic mucosa. These results were insufficient to establish a diagnosis of inflammatory bowel disease (IBD). Nevertheless, we initiated prednisone and salazosulfapyridine according to the standard IBD protocol; however, these treatments were ineffective.
Blood tests on the 60th day revealed liver dysfunction (aspartate transaminase 91 U/L, alanine transaminase 136 U/L, γ-glutamyl transpeptidase 18 U/L, leukocyte alkaline phosphatase 77 U/L, total bilirubin 1.3 mg/dL) and elevated lactic acid and pyruvate. As electron microscopic examination showed increased numbers of mitochondria, we considered the possibility of a mitochondrial disease. Accordingly, we initiated vitamin-and-coenzyme therapy with fursultiamine, riboflavin butyrate, ascorbic acid, tocopherol acetate, biotin, levocarnitine chloride, and ubidecarenone, whereupon the patient’s vomiting disappeared. However, her diarrhea persisted, so we initiated infusion of a combined fat emulsion; we administered it for more than 2 weeks, but eventually had to discontinue it because of deteriorating liver function. We replaced it with Twinline-NF® (EN Otsuka, Hanamaki, Japan; lipid 25%), a liquid enteral nutrition formula that is easy to digest and includes medium-chain triglycerides (MCTs) on the 107th day. After the patient started Twinline-NF® , her diarrhea and liver dysfunction gradually abated, and she was able to transition smoothly from enteral nutrition to oral nutrition. She was discharged from the hospital after 134 days. Later on, immunostaining of her colonic mucosa revealed weak expression of MRC complex I, confirming MRC complex I deficiency (Figures 3a and 3b ) .
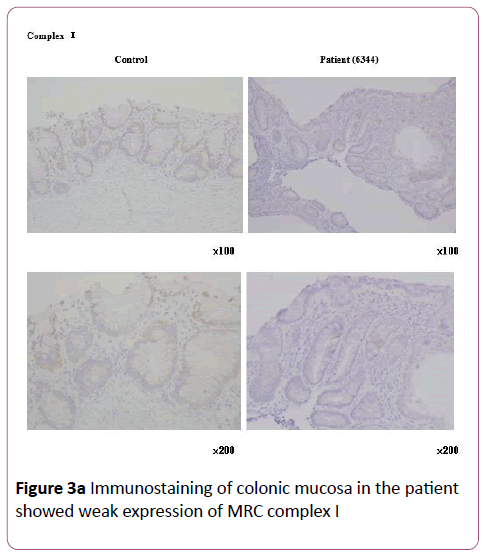
Figure 3a: Immunostaining of colonic mucosa in the patient showed weak expression of MRC complex I.
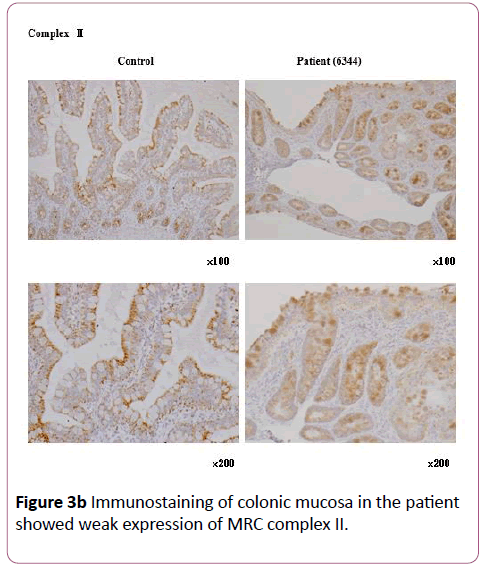
Figure 3b: Immunostaining of colonic mucosa in the patient showed weak expression of MRC complex II.
Discussion
Our patient had gastrointestinal dysfunction caused by low activity of MRC enzyme complex I. At first, we suspected that placing her on total parenteral nutrition had caused her liver dysfunction; however, MRCD was the true cause. We required time to reach the correct diagnosis. MRC complex I deficiency is usually diagnosed by analysis of complex I activity in the diseased organs and tissues (muscle, liver, fibroblasts, etc.). In our patient, it was diagnosed via immunostaining of colonic mucosa collected during colonoscopy. Because the colonic mucosa contains epithelial, muscle, and glandular cells, we did not perform an enzyme activity assay.
Okamura and colleagues were the first to report a case of MRCD associated with gastrointestinal dysfunction, in 1976 [5]; subsequently, other cases were reported from different parts of the world [3,4,6,7]. According to Kawachi and colleagues, about 30% of MRCD patients have gastrointestinal dysfunction, and it is the main manifestation in 13% [8]. The predominant symptoms in such patients are intractable vomiting and/or diarrhea, as in our patient; however, the majority first develop symptoms during the first year of life, and patients such as ours, with symptoms developing after the first year, are infrequently encountered.
Gastrointestinal symptoms have a wide differential diagnosis, so we needed to rule out many different diseases. In addition, because IBD was suspected, we performed a biopsy of the colonic mucosa, whereupon we found nonspecific lymphoid hyperplasia, but not the ulceration typical of IBD. Previous reports of MRCD describe similar findings of nonspecific inflammation. However, as our patient showed no neuropathy or myopathy, we were unable to completely rule out IBD, which delayed arrival at the correct diagnosis. On the other hand, a case has been reported of an infant with MRC complex I deficiency associated with intractable ulcerative colitis, suggesting that even patients with IBD-like ulceration may in fact have MRCD [9].
It has been reported that an increased number of mitochondria and/or mitochondrial swelling on electron microscopy is useful in the diagnosis of MRCD [3]. Our patient indeed showed an increased number of mitochondria (Figure 4), and we recommend that if MRCD is suspected, electron microscopy should be performed on tissue collected from the affected organs.
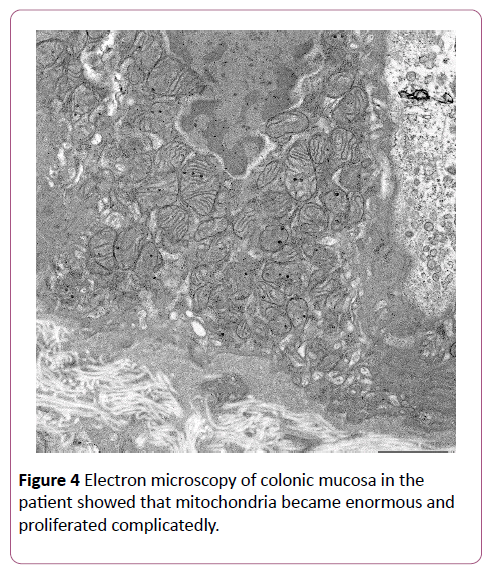
Figure 4: Electron microscopy of colonic mucosa in the patient showed that mitochondria became enormous and proliferated complicatedly.
We believe that the patient’s fever and vomiting episode was not originally due to MRCD. Inadequate excessively low-fat nutrition and increased energy demand due to the episode prompted the worsening of symptoms. As the nutritional treatment for MRCD, a fat-rich diet is appropriate, similar to the treatment for neonatal intrahepatic cholestasis caused by citin deficiency [10]. However, the lipid content of Twinline-NF is similar to that of regular meals. This patient may have not needed a high fat ratio when she was in a state of needing normal energy supply. This might be one of the reasons for no apparent symptoms in infancy.
Accordingly, we initiated infusion of fat emulsion to supply FFAs and stop the patient’s diarrhea; however, because of liver dysfunction, the treatment had to be discontinued before it could take full effect. To decrease the metabolic burden, improve the absorption of fat, and supply MCTs, we switched to nutritional therapy with Twinline-NF®, a liquid enteral nutrition formula containing MCTs. As a result, the patient’s diarrhea episodes decreased in frequency from daily to less than daily, and disappeared entirely within 2 weeks. There have been previous reports of patients treated with MCT formulas, although not with Twinline-NF® [10]. Our results suggest that enteral nutrition formulas containing MCTs can be effective for gastrointestinal symptoms caused by MRCD.
Conclusion
We have reported the case of a 1-year-old girl with MRC complex I deficiency mainly affecting the gastrointestinal tract. This disease should be included in the differential diagnosis for patients with intractable, cryptogenic gastrointestinal symptoms.
Acknowledgments
We thank Masanori Tanaka (Department of Pathology and Laboratory Medicine, Hirosaki Municipal Hospital) and Atsushi Yoden (Department of Pediatrics, Osaka Medical College) for their diagnostic advice.
Author Contributions
Hiroki Kuranobu, Jun Murakami, Naomi Kuranobu and Ken Okamoto collected and analyzed our patient’s data; Kei Murayama performed immunostaining of patient’s colonic mucosa; Hiroki Kuranobu and Jun Murakami wrote the manuscript. Susumu Kanzaki critically reviewed the manuscript and gave conceptual advice. All authors read and approved the final manuscript.
References
- Skladal D, Halliday J, Thorburn DR (2003) Minimum birth prevalence of mitochondrial respiratory chain disorders in children. Brain 126:1905-1912.
- Munnich A, Rustin P (2001) Clinical spectrum and diagnosis of mitochondrial disorders. Am J Med Genet 106: 4-17.
- Teitelbaum JE, Berde CB, Nurko S, Buonomo C, Perez-Atayde AR et al. (2002) Diagnosis and management of MNGIE syndrome in children: Case report and review of the literature. J PediatrGastroenterolNutr 35: 377-383.
- Hirano M, Silvestri G, Blake DM (1994) Mitochondrial neurogastrointestinalencephalomyopathy (MNGIE): Clinical, biochemical, and genetic features of an autosomal recessive mitochondrial disorder. Neurology 44: 721-727.
- Okamura K, Santa T, Nagae K, Omae T (1976) Congenital oculoskeletal myopathy with abnormal muscle and liver mitochondria. J NeurolSci 27: 79-91.
- Nishino I, Spinazzola A, Papadimitriou A (2000) Mitochondrial neurogastrointestinalencephalomyopathy: an autosomal recessive disorder due to thymidine phosphorylase mutations. Ann Neurol 47: 792-800.
- Bardosi A, Creutzfeldt W, DiMauro S (1987) Myo-, neuro-, gastrointestinal encephalopathy (MNGIE syndrome) due to partial deficiency of cytochrome-c-oxidase. A new mitochondrial multisystem disorder. ActaNeuropathol 74: 248-258.
- Kawachi E, Murayama K, Fushimi T (2013) Gastroenterological symptoms of mitochondrial respiratory chain complex deficiency. J PediatrGastroenterolHepatolNutr 27: 148-154.
- Vanderborght M, Nassogne MC, Hermans D (2004) Intractable ulcerative colitis of infancy in a child with mitochondrial respiratory chain disorder. J PediatrGastroenterolNutr 38: 355-357.
- Kaji S, Murayama K, Nagata I (2009) Fluctuating liver functions in siblings with MPV17 mutations and possible improvement associated with dietary and pharmaceutical treatments targeting respiratory chain complex II. Mol Genet Metab 97: 292-296.