Keywords
α-Carbanion/Enamine intermediate; Dimethylallyl pyrophosphate; Isopentenyl pyrophosphate; Non-mevalonate; Site-directed mutagenesis; TPPdependent
Introduction
Isopentenyl pyrophosphate (IPP) and dimethylallyl pyrophosphate (DMAPP) are the precursors for isoprenoids, the largest family of biologically active compounds. The isoprenoid family of molecules represents the most diverse set of biochemical entities known and are of considerable biological importance. For example, cholesterol (an isoprenoid) serves as a precursor to the glucocorticoids, the androgens, the mineralocorticoids, the gestagens, and estrogen [1,2]. The biomedical importance of the isoprenoids has intrigued the scientific community on numerous levels for decades, with one of the earliest questions being: “How these molecules are synthesized in the cell?” This question was answered for cholesterol by Bloch, Cornforth, Lynen, and Popják in the 1930’s through the 1960’s [3-6]. The pathway defined by these researchers from acetyl-CoA to cholesterol is outlined in almost every undergraduate biochemistry textbook. A key intermediate in the canonical pathway of cholesterol and the isoprenoid biosynthesis is mevalonate.
For many years, it was thought that IPP, DMAPP, and all the biologically-occuring isoprenoids were derived solely from the mevalonate-dependent (MVA) pathway. An alternative pathway to IPP, DMAPP, and the isoprenoids was discovered by 1990’s, which accounts for isoprenoid production in the apicomplexan parasites, many eubacteria, green algae, and plants [7]. In this alternative pathway, IPP is not derived from mevalonate, but from pyruvate and glyceraldehyde-3-phosphate. A 5-carbon sugar phosphate, xylulose-5-phosphate is a key intermediate in the mevalonate-independent pathway of isoprenoid biosynthesis [7- 9]. The enzymes catalyzing the reactions of the non-mevalonate dependent pathway (NMVA, also called the mevalonateindependent pathway or MEP) are completely different than the enzymes of the mevalonate dependent pathway (MVA). Many human pathogens produce their isoprenoids exclusively via NMVA or a combination of the MVA and NMVA, including Plasmodium spp, Mycobacterium tuberculosis, Chlamydophila pneumoniae, Treponema pallidum.
The first committed step in the NMVA involves the thiamine pyrophosphate- and divalent metal ion-dependent condensation of pyruvate (Pyr) and D-glyceraldehyde-3-phosphate (GAP) to form 1-deoxy-D-xylulose-5-phosphate (DXP) and CO2; as catalyzed by 1-deoxy-D-ylulose-5-phosphate synthase (DXPS) (Figure 1) [10]. DXP, the product of the DXPS reaction, is used not only for the production of IPP, DMAPP, and the isoprenoids, but also for the biosynthesis of thiamin (vitamin B1) and pyridoxal (vitamin B6) in Plasmodium and other organisms [11-13]. Up- and downregulation of DXPS expression in Arabidopsis thaliana results in the commensurate changes in the level of the isoprenoid levels providing strong evidence that DXPS catalyzes the rate-determing step of NMVA in this organism [14] and, by extension, in others.
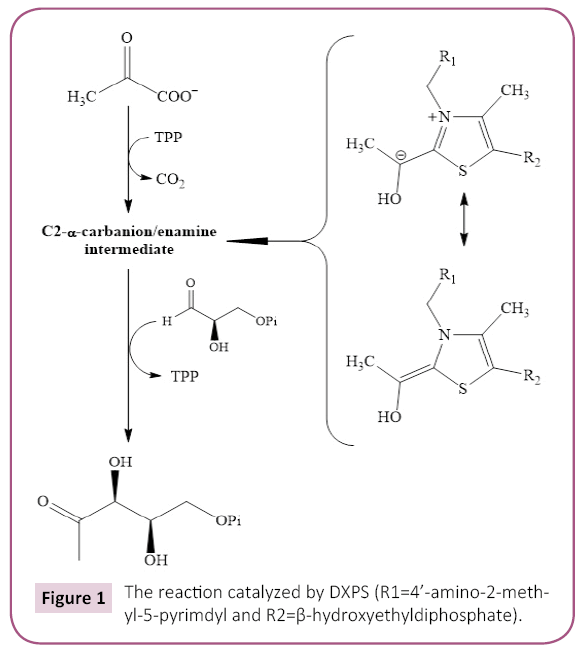
Figure 1: The reaction catalyzed by DXPS (R1=4’-amino-2-methyl- 5-pyrimdyl and R2=ß-hydroxyethyldiphosphate).
The focus of the present study is to define the active site pocket of DXPS from Deinococcus radiodurans by constructing a set of site-directed mutants and to elucidate the order of substrate binding using steady-state kinetic analysis, product inhibition with DXP, and a dead end inhibition with β-fluoropyruvate. This work fosters a better understanding of the DXPS-catalyzed reaction and will aid in the rational design of DXPS inhibitors for the treatment of malaria and other human diseases.
Materials and Methods
Materials
Thiamine pyrophosphate (TPP), pyruvate, D,L-glyceraldehyde-3- phosphate (GAP), 1-deoxy-D-xylulose-5-phosphate sodium salt, bovine serum albumin, and LB-broth were purchased from Sigma Aldrich. NADPH was purchased from Alexis Biochemical, Ni-NTA resin was purchased from Invitrogen, and β-mercaptoethanol (β- Me) was purchased from Fisher. E. coli XL-10 cells, deoxynucleotide mix PCR grade, pfuUltra Hotstart DNA polymerase, QuikChange II site directed mutagenesis kit and acetonitrile (HPLC grade) were purchased from Agilent. The DNA vectors, pET28a (+) and pET15b (+), and E. coli BL-21 B (DE3) cells were purchased from EMD Biosciences. DNA sequencing services and primers were purchased from MWG operon. All the other reagents were of the highest quality commercially available.
Cloning of D. radiodurans DXPS and E. coli 1-Deoxy-D-xylulose 5-phosphate Reductoisomerase (DXR)
A synthetic, codon optimized D. radiodurans dxps gene with 5’- NdeI and 3’-XhoI restriction sites in a pMK vector was purchased from Geneart (Germany). Thedxps gene was excised from the pMK vector and cloned into the NdeI and XhoI sites of a pET28a (+) vector (kanamycin resistance) with N-terminal His6-tag to yield the pET28a (+)-pxps plasmid. Successful cloning of the D. radioduransd xps gene was confirmed by DNA sequencing at MWG Operon.
A synthetic, codon optimized E. coli dxr gene with 5’-NdeI and 3’-BamHI restriction sites in a pMK vector was purchased from Geneart (Germany). The dxr gene was excised from pMK vector and cloned into NdeI and BamHI restriction sites of pET15b (+) vector with a C-terminal His6 tag to yield the pET15b (+)-dxr plasmid. Gene insertion was confirmed by DNA sequencing.
Production of the D. radiodurans DXPS mutants
Site-directed mutagenesis was carried out using the QuikChange II site-directed mutagenesis kit. Briefly, the mutagenesis mixture consists of 50-100 ng plasmid pET28a (+)-dxps as a template, 1X PCR reaction buffer, 0.4 mM each of the forward and reverse primer, 0.25 mM dNTP mixture, 5 μL Quik solution, and 2.5 units of pfu Ultra hot-start polymerase in a 50 μL reaction. The overlap extension method was used to produce the DXPS mutant that was difficult to create via site directed mutagenesis [15]. The sequence of the mutant DNA was confirmed by DNA sequencing.
Over-expression and purification of Wildtype DXPS and the DXPS mutants
Plasmids containing the wild type D. radioduransd xps gene or the mutantdxps gene were transformed into E. coli BL-21 B (DE3) cells and used for protein expression. An overnight culture of E. coli in LB broth containing 50 μg/mL kanamycin was diluted 100- fold, cultured at 37°C until the absorbance at 600 nm reached ~0.6, and then cooled to 20°C. Expression was induced by the addition of 0.5 mM isopropyl β-D-1-thiogalactopyranoside (IPTG). The cells were harvested by centrifugation (6,000 × g for 10 min) after being shaken for 6 hrs at 20°C and the resulting cell pellets stored at -80°C before purification. Cells were thawed and all the purification steps were performed at 4°C. Cells were resuspended in binding buffer (20 mM Tris, 500 mM NaCl, 5 mM imidazole, 10 mM β-mercaptoethanol (β-Me), pH=7.5) supplemented with 1 mM phenylmethanesulfonylfluoride (PMSF), 4 μg/mL leupeptin, and 2 μg/mL pepstatin, sonicated using a Heat systems W-380 ultrasonic processor, and centrifuged (16,000 × g for 20 min) to remove cell debris. The supernatant from the cell lysate was applied to a 1.5 cm × 5 cm column packed with Ni-NTA resin that had been equilibrated with binding buffer. Non-bound proteins eluted from the column by first washing with 5 column volumes of binding buffer followed by 20 column volumes of wash buffer (20 mM Tris pH 7.5, 500 mM NaCl, 60 mM imidazole, and 10 mM β-Me). The bound DXPS (wildtype or mutant) was eluted from the Ni-NTA resin using elution buffer (20 mM Tris pH 7.5, 500 mM NaCl, 250 mM imidazole, and 10 mM β-Me). A flow rate of 1.5 mL/min was maintained through the Ni-NTA column for all the loading and washing steps. DXPS-containing fractions containing were combined, exhaustively dialyzed at 4°C against 20 mM Tris pH 7.5, 100 mM NaCl, and 10 mM β-Me, and concentrated by ultrafiltration. The final yield of DXPS (wildtype or mutant) was 7-8 mg/L of E. coli culture medium. Enzyme was flash frozen in liquid nitrogen, stored at -80°C. The purity of the DXPS (wildtype or the mutant) was evaluated by SDS-PAGE.
Determination of GAP and DXP concentration
D,L-GAP was obtained from Sigma Aldrich as a suspension in water. The concentration of D-GAP was measured spectrophotometrically using glyceraldehyde-3-phosphate dehydrogenase (GAPDH). A reaction mixture (0.5 mL) containing 30 mM sodium pyrophosphate buffer pH 8.6, 5 mM L-cysteine, 0.6 mM NAD+, 0.6 mM sodium arsenate, and D, L-GLP at concentration less than 0.2 mM were incubated at 30°C for 10 min, the reaction initiated with 2 μg of GAPDH (Novus Biotechnology), and the progress of the reaction monitored spectrophotometrically at 340 nm. The final concentration of D-GAP was calculated based on the production of NADPH (ε340=6,220 M-1 cm-1). Throughout this work, the abbreviation “GAP” represents the concentration of D-glyceraldehyde-3-phosphate in the racemic mix.
DXP was purchased from Sigma Aldrich as the sodium salt. The salt was resuspended in 10 mM HEPES buffer (pH=6.0), and the concentration was measured spectrophotometerically using DXR. A reaction mixture (0.5 mL) containing of 100 mM HEPES pH 8.0, 1 mg/ml BSA, 1.5 mM MnCl2, 0.3 mM NADPH, and DXP at concentration less than 0.1 mM were incubated at 37°C for 5 min, the reaction initiated with 1.25 μg of DXR, and the progress of the reaction monitored spectrophotometrically at 340 nm. Final concentration of DXP was calculated based on the oxidation of NADPH (ε340=6,220 M-1 cm-1).
Assays for DXPS activity
One assay we employed to measure DXPS activity involved a DXPSDXR coupled assay. In this way, the DXPS-dependent production of DXP is ultimately coupled to the oxidation of NADPH to NADP+. The solution for the DXPS-DXR coupled contained 100 mM HEPES pH 8.0, 100 mM NaCl, 1 mg/ml BSA, 1 mM TPP, 1.5 mM MnCl2, 2 mM β-Me, 0.15 mM NADPH, 0.2 mg/ml DXR, and varying concentrations of pyruvate or GAP [16]. Steady-state kinetic experiments were performed by varying pyruvate or GAP at a fixed saturating concentration of the co-substrate. A DXPS-DXR reaction solution was incubated at 37°C for 5 min, the reaction was initiated by addition of 358 nM DXPS, and the progress of the reaction monitored spectrophotometrically at 340 nm for the oxidation of NADPH.
Another assay we employed to measure DXPS activity was an HPLC determination of net [DXP] production or [GAP] consumption, modified from a published procedure [17]. This endpoint assay to measure DXPS activity was used in our product inhibition studies. Briefly, the reaction was initiated by addition of 573 nM DXPS. At the desired time, an aliquot (50 μL) was removed and added to an equal volume of acidic 2,4-dinitrophenol (DNP) (100 mM 2,4- DNP in 2N H2SO4) to terminate the enzymatic reaction. After a 5 minute incubation at room temperature, the pH was adjusted to 5-7 by addition of 36 μL 4.0 M Tris buffer pH 10.0 and centrifuged to remove excess 2,4-DNP. The DNP derivatives of pyruvate, GAP, and DXP were separated using a 5 μm Discovery® C18 column (25 cm × 4.6 mm) developed using the following conditions: flow rate=1.5 mL/min; solvent A, 100 mM ammonium acetate pH 4.6, 0.05% TFA; solvent B, acetonitrile, 0.05% TFA; 20% - 35% solvent B over 12 min; and then 35–60% solvent B over 2 min. When varying [pyruvate] at one fixed [GAP], the concentrations used for the initial velocity kinetic experiments were as follows: 1.0 mM GAP and pyruvate at 0.26, 0.50, 0.75, or 4.0 mM; and DXP at 0, 0.2, 0.3, or 0.4 mM. The DXP hydrazone peak area was measured and the DXP concentration was determined against a standard curve generated using DXP hydrazone. When carrying the DXP inhibition studies, net [DXP] produced was determined by substrating background [DXP] at zero time (before the addition of enzyme) from the total [DXP] produced at the desired time interval. When varying [GAP] at one fixed [pyruvate], the concentrations used for the initial velocity kinetic experiments were as follows: 0.3 mM pyruvate and GAP at 0.05, 0.1, 0.25, and 0.50 mM; and DXP at 0, 0.2, 0.3, or 0.4 mM. The GAP hydrazone peak area was measured and the GAP concentration was determined compared against a standard curve generated using GAP hydrazone. Assays were performed in duplicate.
Inhibition of DXPS by β-fluoropyruvate (F-Pyr)
Initial reaction velocities were determined using the DXPS-DXR coupled assay. When varying [pyruvate] at one fixed [GAP], the concentrations used for the initial velocity kinetic experiments were as follows: 0.5 mM GAP, pyruvate, 0.1 mM, 0.18 mM, 0.5 mM, or 2 mM; and F-Pyr at 0 μM, 25 μM, 50 μM, 75 μM or 100 μM. When varying [GAP] at one fixed [pyruvate], the concentrations used for the initial velocity kinetic experiments were as follows: 2.0 mM pyruvate and GAP at 0.025 mM, 0.05 mM, 0.125 mM, or 0.25 mM; and F-Pyr at 0, 25 mM, 50 mM, 75 mM or 100 μM. Assays were performed in triplicate using 115 nM of D. radiodurans DXPS and the data were fit using nonlinear least-squares regression analysis.
Analysis of the initial velocity kinetic data
The steady state initial velocity for DXPS measured at various concentrations of pyruvate and GAP were fit to equation 1 using nonlinear regression analysis in Sigma-Plot 12.0 Data for inhibition of DXP and fluoropyruvate against pyruvate and GAP were fit to equation 2, 3 and 4 for competitive, noncompetitive, and uncompetitive, respectively, where [I] is the concentration of inhibitor and Ki is the inhibition constant with respect to β-fluoropyruvate.
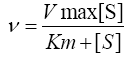 (1)
(1)
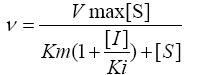 (2)
(2)
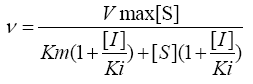 (3)
(3)
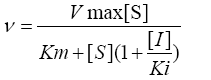 (4)
(4)
Results and Discussion
Overexpression and purification of D. radiodurans DXPS
A synthetic, codon-optimized wildtype D. radioduransd xps gene (dxps-wt) with a N-terminal His6 tag was cloned into the pET28a (+) vector using NdeI and XhoI restriction sites. A series of D. radiodurans DXPS mutant proteins were constructed near the active site pocket using thedxps -wt gene as template by the Quikchange site-directed mutagenesis method or the overlap extension method [15]. Wildtye enzyme (DXPS-WT) and the mutant enzymes (all possessing N-terminal His6 tag) were affinity purified using the Ni-NTA resin to >95% pure with respectable expression yields of 6-8 mg per liter of culture.
Comparison of steady-state kinetic parameters between mutant DXPS proteins and wildtype enzyme
We have constructed and evaluated a set of mutant D. radiodurans DXPS proteins. DXPS is a TPP- and Mg (II)-dependent enzyme that belongs to the transketolase (TK) family of TPPdependent enzymes [18,19]. The sequences of E. coli and D. radiodurans DXPS are 45% identical and their respective structures are remarkably similar. The rms distance of equivalent Cα atoms between the two structures is only 0.7Å [19]. Despite low sequence identity to other TK family members, E. coli and D. radiodurans structures are similar to that of yeast transketolase (TK), the E1 subunit of E. coli pyruvate dehydrogenase (PDH), and P. putida 2-oxoisovalerate dehydrogenase [19,20]. DXPS is a dimer, with each monomer possessing three domains: domains I, II, and II (Figure 2). Domain I is composed of residues 1-319, domain II is composed of residues 320-495, and domain III is composed of residues 496-629. The DXPS active site is found at interface of domains I and II within the same monomer (Figures 3 and 4) [19,20]. In constrast to DXPS, the active site of other TPP-dependent enzymes is located at the interface of domain I of one monomer and domain II of the second monomer, examples include the E1 component of E. coli pyruvate dehydrogenase [21], yeast transketolase [22], yeast acetohydroxyacid synthase [23], Bifidobacterium longum phosphoketolase [24]. TPP is bound deeply within a cleft formed from segments of domain I and II and adopts a V-conformation, like that found in other TPP-dependent enzymes [19]. The V-conformation of TPP positions the 4’-imino group to deprotonate the C2 atom, thus, forming the catalytically important C2 carbanion [25,26]. Sequence alignments of DXPS proteins from a number of different organisms show extensive sequence identity at the binding sites for TPP and GAP [19,20].
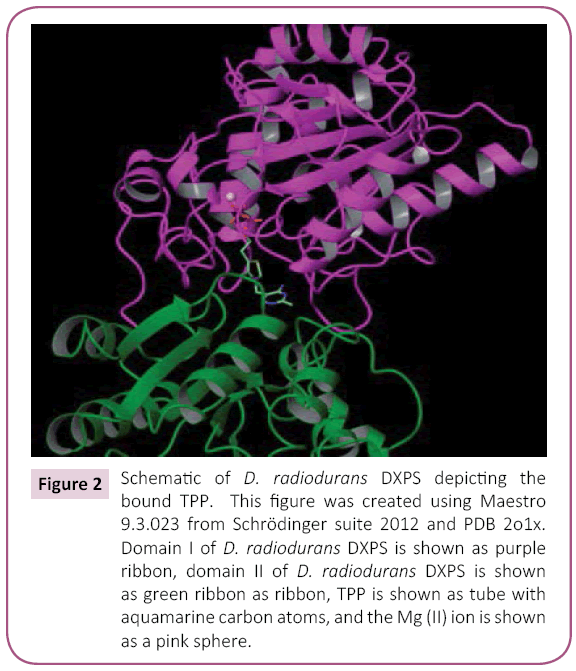
Figure 2: Schematic of D. radiodurans DXPS depicting the bound TPP. This figure was created using Maestro 9.3.023 from Schrödinger suite 2012 and PDB 2o1x. Domain I of D. radiodurans DXPS is shown as purple ribbon, domain II of D. radiodurans DXPS is shown as green ribbon as ribbon, TPP is shown as tube with aquamarine carbon atoms, and the Mg (II) ion is shown as a pink sphere.
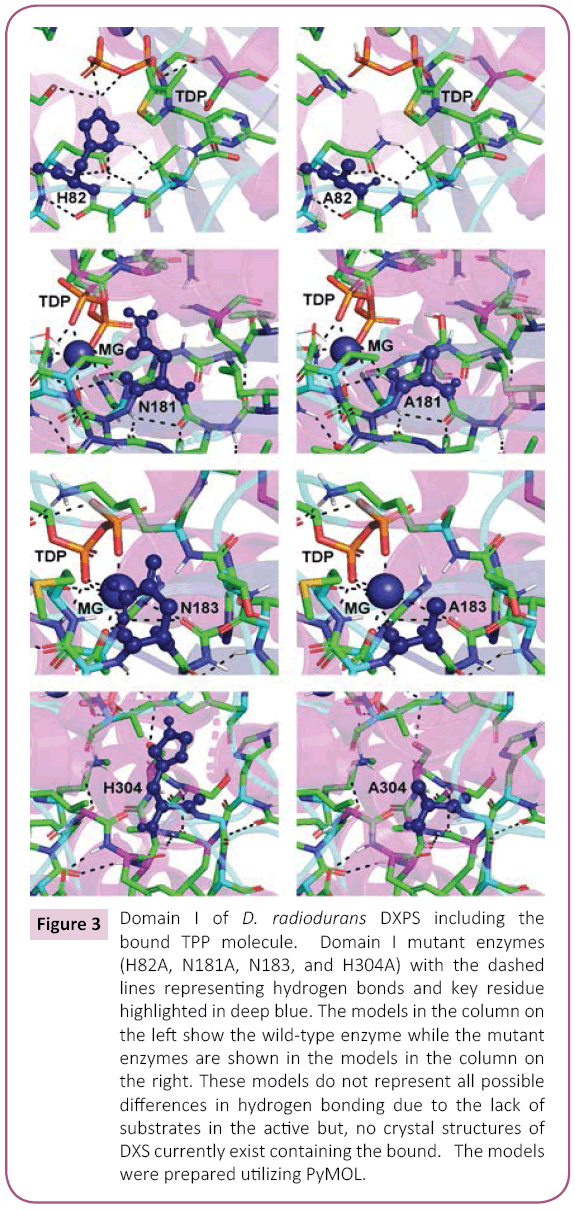
Figure 3: Domain I of D. radiodurans DXPS including the bound TPP molecule. Domain I mutant enzymes (H82A, N181A, N183, and H304A) with the dashed lines representing hydrogen bonds and key residue highlighted in deep blue. The models in the column on the left show the wild-type enzyme while the mutant enzymes are shown in the models in the column on the right. These models do not represent all possible differences in hydrogen bonding due to the lack of substrates in the active but, no crystal structures of DXS currently exist containing the bound. The models were prepared utilizing PyMOL.
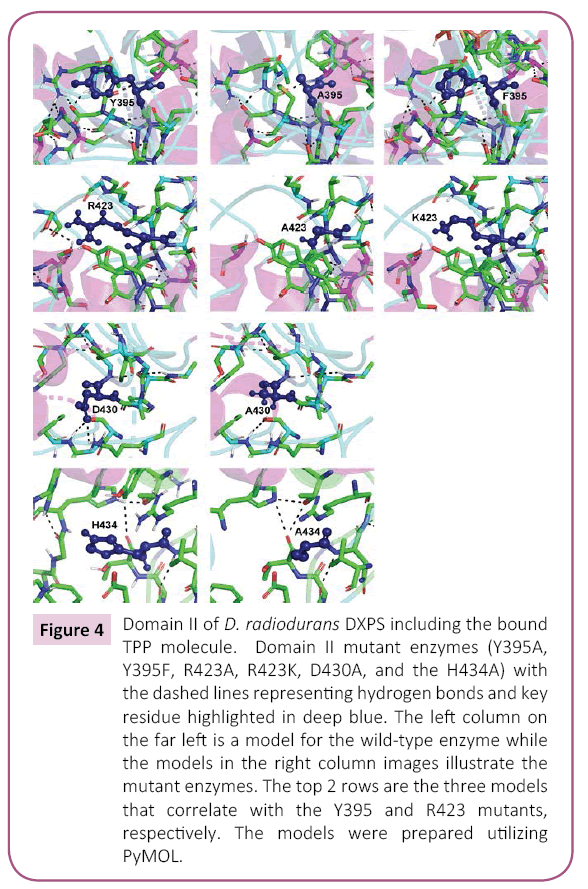
Figure 4: Domain II of D. radiodurans DXPS including the bound TPP molecule. Domain II mutant enzymes (Y395A, Y395F, R423A, R423K, D430A, and the H434A) with the dashed lines representing hydrogen bonds and key residue highlighted in deep blue. The left column on the far left is a model for the wild-type enzyme while the models in the right column images illustrate the mutant enzymes. The top 2 rows are the three models that correlate with the Y395 and R423 mutants, respectively. The models were prepared utilizing PyMOL.
Domain I mutants
A structural feature common too many TPP-dependent enzymes is the presence of three discrete domains, as was found for DXPS. The Pyr domain interacts with the pyrimidine ring of the TPP cofactor and PP domain interacts with the pyrophophosphate tail of TPP. The function of the third domain is unclear, but may serve a regulatory function as this domain is structurally related to nucleotide binding domains in other proteins [27]. As reviewed by Duggleby [27], the relative arrangement of the three domains is different within the various families of the TPP-dependent enzymes. Domain I of D. radiodurans DXPS is the PP domain and functions primarily in the binding of the pyrophosphate moiety of TPP. The DXPS-bound Mg (II) is coordinated to the TPP pyrophosphate, meaning the amino acids necessary for Mg (II) binding are found within domain I. GAP is bound within a pocket that is composed of amino acids from both domains I and II; thus, domain I also has a role in GAP binding.
The results for the domain I mutants constructed herein are shown in Tables 1 and 2. His-82 and His-304 are two amino acid residues involved in TPP binding: the N3 imidazole His-82 is 5.6Å from C2 of the thiazolium ring and the N1 of the imidazole of His-304 is 5.2Å from the C2 of the thiazolium ring. Also, the N1 of the imidazole is 4.1Å from N1 of the imidazole of His-304 in D. radiodurans DXPS. The mutation of both to Ala, generating separate H82A and H304A mutant proteins, yielded catalytically defective enzymes with Vmax and (V/K)app values of 2-12% of wild-type. Given the proximity of His-82 and His-304 in the D. radiodurans DXPS structure, it seems likely that these amino acids could function “in reserve” of each other accounting for the ~10% remaining in both H82A and H304A mutants. In other words, His-304 can partially fulfill the role played by His-82 in catalysis after its mutation to Ala.
| Protein |
KM,app
(mM) |
kcat,app
(s-1) |
(kcat/KM)app
(s-1 M-1) |
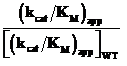 |
| Wildtype |
0.28 ± 0.03 |
7.4 ± 0.3 |
2.6 × 104 |
1.0 |
| H82A |
0.23 ± 0.02 |
0.38 ± 0.01 |
1.7 × 103 |
0.07 |
| N181A |
0.17 ± 0.02 |
3.3 ± 0.1 |
1.9 × 104 |
0.7 |
| N183A |
0 |
0 |
0 |
0 |
| H304A |
1.7 ± 0.5 |
0.90 ± 0.01 |
5.8 × 102 |
0.02 |
aApparent kinetic constants were measured by varying the initial concentration of pyruvate at an initial fixed concentration of 0.5 mM GAP.
Table 1: Domain I mutants: Measurement of steady-state kinetic parameters with GAP is the variable substratea.
| Protein |
KM,app
(mM) |
kcat,app
(s-1) |
(kcat/KM)app
(s-1 M-1) |
|
| Wildtype |
0.05 ± 0.01 |
7.9 ± 0.4 |
1.5 × 105 |
1.0 |
| H82A |
0.03 ± 0.01 |
0.37 ± 0.02 |
1.3 × 104 |
0.09 |
| N181A |
0.03 ± 0.01 |
0.43 ± 0.02 |
1.4 × 104 |
0.09 |
| N183A |
0 |
0 |
0 |
0 |
| H304A |
0.08 ± 0.02 |
0.90 ± 0.1 |
1.1 × 104 |
0.07 |
aApparent kinetic constants were measured by varying the initial concentration of GAP at an initial fixed concentration of 2.0 mM pyruvate.
Table 2: Domain I mutants: Measurement of steady-state kinetic parameters with pyruvate is the variable substratea.
The KM,app values for the H82A mutant for both GAP and pyruvate are similar to wildtype indicating that His-82 contributes little to the binding of either substrate. Similar to our results for the H82A mutant, mutation of the equivalent amino acid in yeast transketolase, His-69, to Ala yielded an enzyme with a Vmax that is 1.5% of wildtype and virtually the same KM for the acceptor substrate (ribose 5-phosphate for transketolase and GAP for DXPS). In contrast to our results for H82A, the donor substrate KM in yeast transketolase does increase 6-fold [28]. These data indicate that His-82 and His-69 serve to stabilize the α-carbanion/enamine-TPP intermediate produced from the donor substrate in DXPS and transketolase, which was observed later in the structure of the yeast transketolase-α, β-dihydroxyethyl-TPP complex [29]. One difference between the two enzymes is the role served by His-82 or His-69 in donor substrate binding. In D. radiodurans DXPS, His-82 seems to have no role in donor substrate binding while in yeast transketolase, His-69 does contribute to donor substrate binding.
The results for the H304A mutant differed from that of H82A as the KM,app for GAP was similar to wildtype, but the KM,app value for pyruvate was 6-fold higher than wildtype. The structure of D. radiodurans DXPS shows that His-304 is located within an open cavity near the thiazolium C2 atom and, thus, likely contributes to both pyruvate binding and stabilization of the α-carbanion/ enamine-TPP intermediate.
Other mutations that were constructed within domain I were N181A and N183A. Asn-183 is coordinated to the bound Mg (II) ion and mutation of this residue to Ala results in a completely inactive protein. In contrast to N183A, the N181A mutant retains activity exhibiting 10% of (V/K)app,GAP and 70% of (V/K)app,pyruvate. The KM,app values for both substrates are approximately the same as wildtype for the N181A mutant; thus, the decrease in (V/K)app reflects a decrease in catalytic efficiency upon mutant of Asn181 to Ala. The amino acid equivalent to Asn-181 in yeast transketolase is Asp-185. The structure of yeast transketolase showns that Asp-185 is involved in a network of bound water molecules important in Ca (II) chelation and pyrophosphate binding in this enzyme. Asn-181 may serve a similar role in D. radiodurans DXPS. A mutation in this residue could subtly alter the active site architecture such that the TPP is no longer optimally positioned for catalysis. Models of the domain I mutants illustrating the critical active site amino acids and hydrogen bonds are shown in Figure 3.
Domain II mutants
Amino acids 320-495 of D. radiodurans DXPS define domain II, which primarily interacts with the thiazolium and aminopyrmidine rings of TPP. Thus, domain II in DXPS is the Pyr domain [27]. Domain II also serves an important role in the binding of both substrates, GAP and pyruvate [19,20]. Of the amino acids in domain II that we altered via mutagenesis, the largest effects on catalysis were observed in changing Arg-423 (Tables 3 and 4). Arg-423 is conserved in other DXPS enzymes and has a role in GAP binding resulting from the formation of hydrogen bond between its guanidino group and the phosphate of GAP. Mutation of Arg-423 to a lysine yields an enzyme with considerable residual activity (50-80%), but with altered KM values; the KM ,GAP increasing 12-fold and the KM ,pyruvate decreasing 5-fold. Mutation of Arg-423 to an alanine yields a mutant enzyme with little activity, a (V/K) app that is 0.1% of wildtype. The KM ,GAP for the R423A mutant is approximately equivalent to the KM for glyceraldehyde in DXPS WT (14 mM), providing additional evidence that the guanidino moiety of Arg-423 interacts with the phosphate of GAP. The amino acid equivalent to Arg-423 in D. radiodurans DXPS in yeast transketolase is His-469. The structure of yeast transketolase shows that imidazole of His-469 forms a hydrogen bond with the phosphate of the acceptor substrate (erythrose 4-phosphate) [30] and, in addition, may form a similar hydrogen bond to the phosphate in the substrate-TPP adduct [31]. In contrast to the R423 mutant described herein, the yeast transketolase H469A mutant retains considerable activity, but exhibits ~10-fold higher KM values for both substrates. While Arg-423 and His-469 have key roles in the binding of the phosphorylated substrates in both DXPS and transketolase, the positive charge of Arg-423 is more important to catalysis in DXPS than that of His-469 in transketolase.
| Protein |
KM,app
(mM) |
kcat,app
(s-1) |
(kcat/KM)app
(s-1 M-1) |
|
| Wildtype |
0.05 ± 0.01 |
7.9 ± 0.4 |
1.5 × 105 |
1.0 |
| Y395A |
0.67 ± 0.05 |
3.9 ± 0.1 |
5.8 × 103 |
0.04 |
| Y395F |
0.59 ± 0.04 |
5.9 ± 0.2 |
1.0 × 104 |
0.07 |
| D430A |
0.12 ± 0.01 |
7.7 ± 0.2 |
6.6 × 104 |
0.4 |
| R423A |
12 ± 3.2 |
1.7 ± 0.3 |
1.4 × 102 |
9 × 10-4 |
| R423K |
0.60 ± 0.1 |
6.4 ± 0.2 |
1.1 × 104 |
0.07 |
| H434A |
0.23 ± 0.01 |
9.6 ± 0.3 |
4.2 × 104 |
0.3 |
aApparent kinetic constants were measured by varying the initial concentration of GAP at an initial fixed concentration of 2.0 mM pyruvate.
Table 3: Domain II mutants: Measurement of steady-state kinetic parameters with GAP is the variable substratea.
| Protein |
KM,app
(mM) |
kcat,app
(s-1) |
(kcat/KM)app
(s-1 M-1) |
|
| Wildtype |
0.28 ± 0.03 |
7.4 ± 0.3 |
2.6 × 104 |
1.0 |
| Y395A |
0.16 ± 0.01 |
3.6 ± 0.1 |
2.2 × 104 |
0.8 |
| Y395F |
0.19 ± 0.01 |
2.9 ± 0.02 |
1.6 × 104 |
0.6 |
| D430A |
0.52 ± 0.03 |
7.2 ± 0.2 |
1.4 × 104 |
0.5 |
| R423A |
NDb |
NDb |
NDb |
NDb |
| R423K |
0.06 ± 0.01 |
3.9 ± 0.1 |
6.5 × 104 |
2.5 |
| H434K |
1.7 ± 0.1 |
9.9 ± 0.2 |
5.9 × 103 |
0.2+ |
aApparent kinetic constants were measured by varying the initial concentration of GAP at an initial fixed concentration of 0.5 mM GAP.
bND = Not Determined
Table 4: Domain II mutants: Measurement of steady-state kinetic parameters with pyruvate is the variable substratea.
Modeling studies suggest that the hydroxyl group of Tyr-395 interacts directly with the phosphate of GAP, yet it has been reported that the mutation of the equivalent tyrosine residue in E. coli DXPS, Tyr-392, has little effect on catalysis [19]. Both the Y392A and Y392F mutants of E. coli DXPS are more active than wildtype enzyme. We find that mutation of Tyr-395 in D. radiodurans DXPS does compromise catalytic efficiency as both the Y395A and Y395F mutant exhibit kcat/KM values that are only >10% of that of wildtype (Table 1) for GAP. The decrease in catalytic efficiency in both the Y395A and Y395F mutants is largely reflected in a >10-fold increase KM for both proteins. The Y395F mutant is a better catalyst than the Y395A mutant, (kcat/ KM )Y395F/ (kcat/KM)Y395A=1.7, indicating that the aromatic ring and hydroxyl group of Y395 contribute positively to catalysis. This is consistent with studies of the amino acid equivalent of Tyr-395 in yeast transketolase, Phe-442, showing that aromatic side chain of Phe-442 contributes to a hydrophobic binding pocket for the pyridine ring of the TPP cofactor [31].
Like Tyr-395, Asp-430 is thought to interact with GAP, in this case, forming a hydrogen bond to the hydroxyl group at carbon-2. The equivalent amino acid in yeast transketolase, Asp-477, similarly forms a hydrogen bond to the 2-hydroxyl group of erythrose 4-phosphate [30]. Mutagenesis of Asp-430 to alanine yields a mutant enzyme that retains considerable activity, with a V/K that is ~50% of wildtype, resulting from a ~2-fold increase in the KM for both substrates (Table 1). Construction of the similar mutant in yeast transketolase, D477A, yielded a protein of much lower activity, 1-2% of wildtype, with relatively greater values for the substrates. The KM for the donor substrate in the D477A mutant, xylulose 5-phosphate, increased 9-fold while that for the acceptor substrate, ribose 5-phosphate, increased 12-fold [30]. In the reaction catalyzed by DXPS, pyruvate is the donor substrate and GAP is the acceptor substrate. These data suggest that Asp-477 in yeast transketolase has a more important role in acceptor substrate binding and catalysis that Asp-430 in D. radiodurans DXPS.
The amino acid equivalent of His-434 in D. radiodurans DXPS is His-481 in yeast transketolase and Gln-428 in human transketolase [32]. Mutation of Gln-428 to Ala, His, Asn, or Glu in human transketolase yields an enzyme that retains activity with the Vmax values ranging from 5% of wildtype for Q428E to 18% of wildtype for Q428N. More detailed analysis Q428 shows no difference in the KM values relative to wildtype for either the donor or acceptor substrates, but with a decrease in Vmax to 15% of wildtype. These results suggested that Gln-428 was not important to substrate binding in human transketolase, but was important to catalysis. Singleton et al. suggested that Gln- 428 aids in orienting an active site water molecule critical to the optimum positioning of TPP [32]. Mutagenesis studies of His-481 in yeast transketolase differed from what was found for Gln-428 in the human enzyme. Conversion of His-481 to Ala, Ser, or Gln produced of lower activity, with the Vmax values all ≤ 5% of wildtype. Steady-state kinetic analysis of the yeast transketolase mutants showed no effect on the KM for the acceptor substrate, but a large increases in the KM value for the donor substrate, ranging from an 18-fold increase for H481A to 58-fold for the H481Q [28]. Crystallographic studies of yeast transketolase in the presence of β-hydroxypyruvate only, which will form the enzyme bound α,β-dihydroxyethyl-thiamin intermediate, shows that His- 481 interacts with the donor substrate and forms a stabilizing hydrogen bond from the N1 of the imidazole ring to the C2 hydroxyl of this intermediate [29].
Analysis of the H434A mutant of D. radiodurans DXPS yielded results that differed from what has been reported for human and yeast transketolases. The Vmax of the H434 mutant is higher than that of wildtype, Vmax (H434)/Vmax (wildtype)=1.2 - 1.3, and the KM values for both the donor and substrates in the H434 mutant are also increased approximately the same relative to wildtype. Mutagenesis of the equivalent amino acid in E. coli DXPS, His-431, to alanine produced an enzyme with the same activity as wildtype [19], consistent with our data on the D. radiodurans enzyme. The KM for GAP increased 4-fold while the KM for pyruvate increased 6-fold (Table 3). Modeling studies of D. radiodurans DXPS suggest that His-434 is located with the GAP binding pocket without an obvious direct interaction with the substrate. The negatively charged phosphate of GAP seems to dock into a positively charged well in the active site and His-434 seems to contribute to this positively charged active site well. Based on the model of pyruvate-TPP adduct in D. radiodurans DXPS, it is conceivable that pyruvate may also interact with this postively charged well in the active site. Replacement of His-434 with alanine will decrease the net positive charge of the well and increasing the KM for both substrates with little effect on the rate of catalysis. Models of the domain II mutants illustrating the critical active site amino acids and hydrogen bonds are shown in Figure 4.
Inhibition and kinetic studies
The kinetic mechanism has been investigated for many TPPdependent enzymes; a classic ping-pong scheme is common within this family of enzymes [33-35]. A ping-pong kinetic mechanism means that first substrate binds followed by the release of the first product before the second substrate binds. DXPS is an unusual TPP-dependent enzyme as substrates, pyruvate and GAP, bind before pyruvate is decarboxylated; thus, the kinetic mechanism for DXPS is not ping-pong [36-39]. Consistent with this unusual aspect of DXPS-mediated catalysis is the incredible stability of the C2-α-lactyl-TPP intermediate within the active site [38]. The binding of GAP to form the E•C2-α-lactyl- TPP•GAP ternary complex triggers CO2 release, yielding the C2- α-carbanion/enamine that will react with GAP to produce DXP [38,39].
Dead-end inhibition studies and 14CO2 trapping experiments demonstrated that the Rhodobacter encapsulates DXPS has an ordered sequential mechanism with pyruvate binding first [36]. Eubanks and Pouter [36] found that the conversion of pyruvate to 14CO2 is inefficient in absence of GAP, consistent with stability of the DXPS-bound C2-α-lactyl-TPP intermediate. Studies of E. coli DXPS showed that the kinetic mechanism for this enzyme was rapid equilibrium random; pyruvate and GAP binding to E. coli DXPS is reversible and completely independent of each other [37-39].
In our study of D. radiodurans DXPS, we employed a set of steadystate kinetic experiments by varying the concentration of one substrate and at constant, but different concentrations of the other substrate. Results from these experiments were coupled with product (DXP) inhibition data and dead-end inhibition data (β-fluoropyruvate) to provide information regarding the kinetic mechanism. Double reciprocal analysis at lower concentrations of pyruvate (0.02 mM - 0.06 mM) (Figure 5A) and GAP (0.01 - 0.03 mM) (Figure 5B) at different, but fixed concentrations of the other substrate revealed a non-competitive pattern with change in both slope and intercept, eliminating a ping-pong and rapid equilibrium ordered kinetic mechanism [40]. The lack of curvature in the plots at high substrate concentration further suggests that the kinetic mechanism for D. radiodurans DXPS is not steady-state random [40,41].
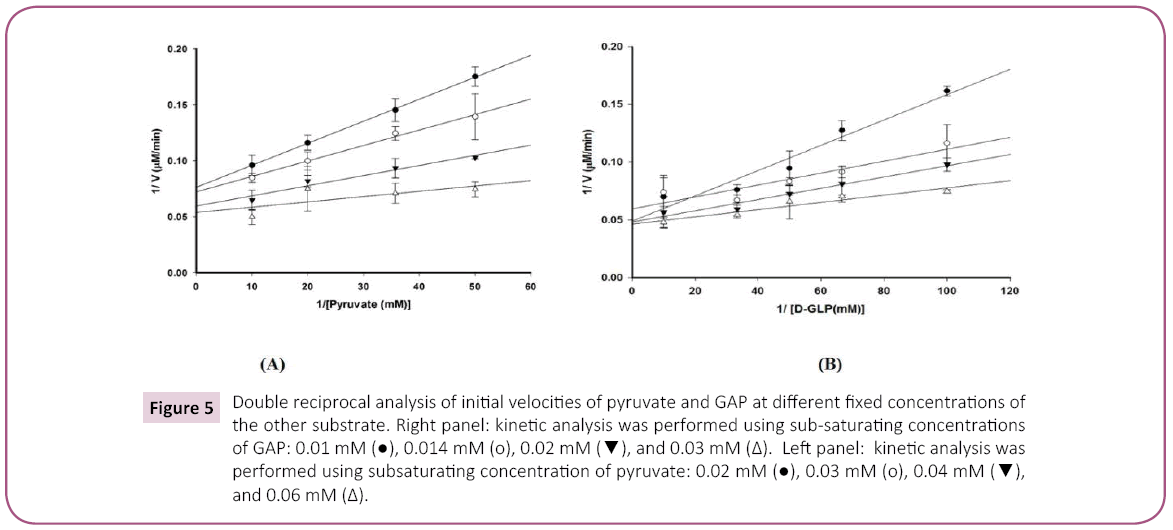
Figure 5: Double reciprocal analysis of initial velocities of pyruvate and GAP at different fixed concentrations of the other substrate. Right panel: kinetic analysis was performed using sub-saturating concentrations of GAP: 0.01 mM ( ), 0.014 mM (o), 0.02 mM (
), 0.014 mM (o), 0.02 mM ( ), and 0.03 mM (?). Left panel: kinetic analysis was performed using subsaturating concentration of pyruvate: 0.02 mM (), 0.03 mM (o), 0.04 mM (), and 0.06 mM (?).
), and 0.03 mM (?). Left panel: kinetic analysis was performed using subsaturating concentration of pyruvate: 0.02 mM (), 0.03 mM (o), 0.04 mM (), and 0.06 mM (?).
Product inhibition studies were next used to determine the mechanism of substrate binding and product release in the D. radiodurans DXPS catalyzed reaction. DXP was used as product inhibitor against pyruvate and GAP. We found that DXP is a weak inhibitor with ~60% inhibition at concentration of 0.4 mM, yielding a Ki in low millimolar range (Table 5). DXP showed a competitive pattern vs. pyruvate indicating that DXP competes for the same enzyme form as pyruvate (Figure 6A), despite the chemical resemblance between DXP and GAP. When GAP was the varied substrate at fixed sub-saturating concentration of pyruvate, DXP yielded a non-competitive inhibition pattern (Figure 6B). This shows that DXP and GAP do not bind to the same enzyme form. At a saturating concentration of pyruvate, no inhibition was detected by DXP when GAP was varied substrate. On the other hand, DXP inhibition was still observed at saturating concentration of GAP when pyruvate was the variable substrate. F-Pyr was used as a dead-end inhibitor to further study the order of substrate binding in D. radiodurans DXPS. F-Pyr is competitive vs. pyruvate (Figure 7A) and non-competitive vs. GAP (Figure 7B) and exhibits a Ki =3.3 μM when pyruvate was the variable substrate. In sum, all the data presented herein, (a) the DXP inhibition patterns, (b) the double reciprocal plots obtained in varying both substrates, and (c) the F-Pyr inhibition point to either a Theorell-Chance ordered, steady-state ordered, or rapid equilibrium random kinetic mechanism. A Theorell- Chance kinetic mechanism, a scheme that requires the lack of a kinetically significant E•C2-α-lactyl-TPP•GAP ternary complex, seems unlikely given the results of Brammer-Basta et al. [39] for E. coli DXPS and is not commonly found as a kinetic mechanism for TPP-dependent enzymes. Mostly likely kinetic mechanism for D. radiodurans DXPS is rapid equilibrium random. Note that a random mechanism could evolve to an ordered mechanism at high [pyruvate], similar to that reported for R. capsulatus DXPS [36]. At high [pyruvate], the D. radiodurans DXPS-catalyzed conversion of pyruvate and GAP to DXP and CO2 would proceed preferentially through path A relative to path B (Figure 8).
| Inhibitor |
Substrate |
Pattern |
Ki (mM) |
| DXP |
Pyruvate |
Competitive |
130 ± 32 |
| DXP |
GAP |
Non-competitive |
830 ± 160 |
| F-Pyr |
Pyruvate |
Competitive |
3.3 ± 0.4 |
| F-Pyr |
GAP |
Non-competitive |
57 ± 3.2 |
Table 5: Inhibition patterns and inhibition constants for the inhibition of D. radiodurans DXPS by DXP or β-fluoropyruvate.
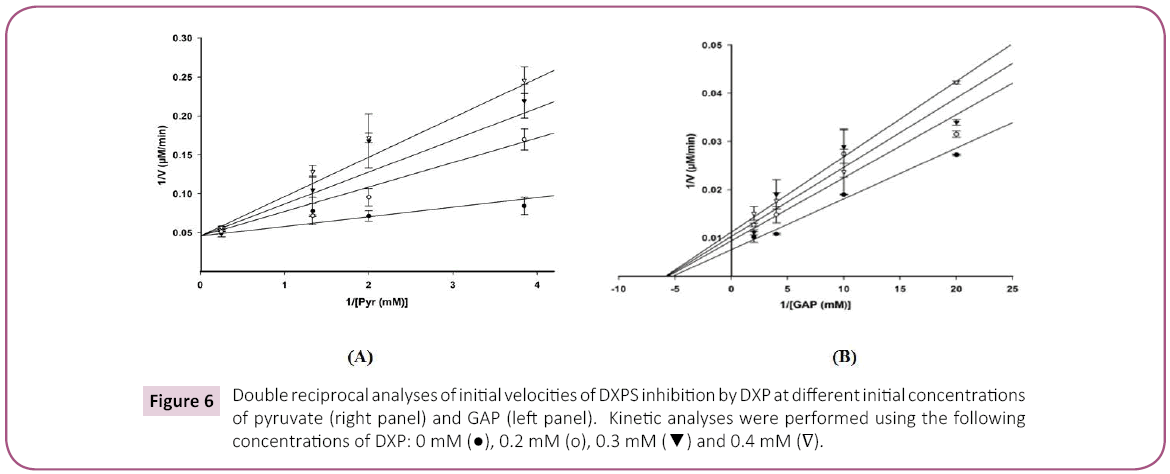
Figure 6: Double reciprocal analyses of initial velocities of DXPS inhibition by DXP at different initial concentrations of pyruvate (right panel) and GAP (left panel). Kinetic analyses were performed using the following concentrations of DXP: 0 mM (), 0.2 mM (o), 0.3 mM () and 0.4 mM ( ).
).
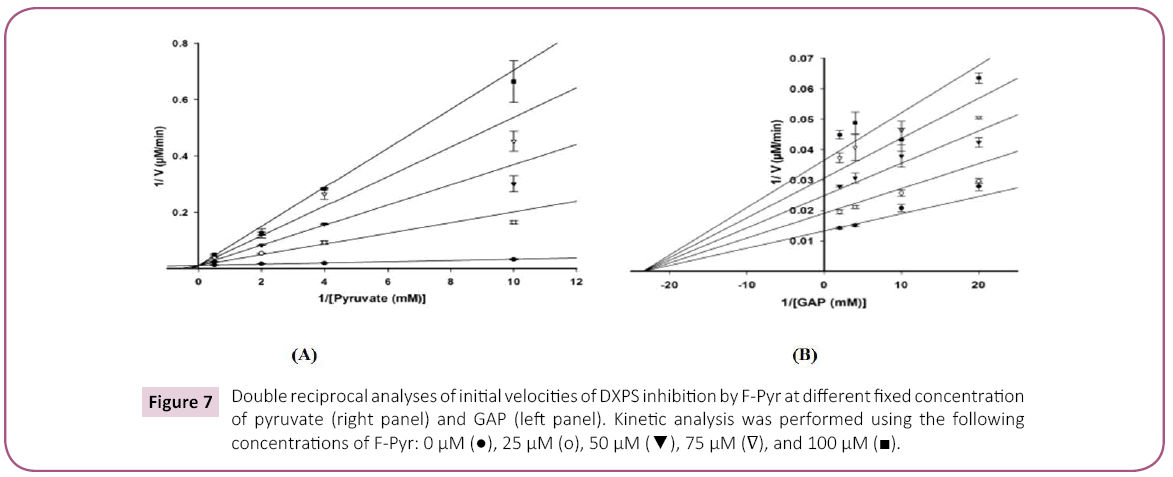
Figure 7: Double reciprocal analyses of initial velocities of DXPS inhibition by F-Pyr at different fixed concentration of pyruvate (right panel) and GAP (left panel). Kinetic analysis was performed using the following concentrations of F-Pyr: 0 µM (), 25 µM (o), 50 µM (), 75 µM (?), and 100 µM ( ).
).
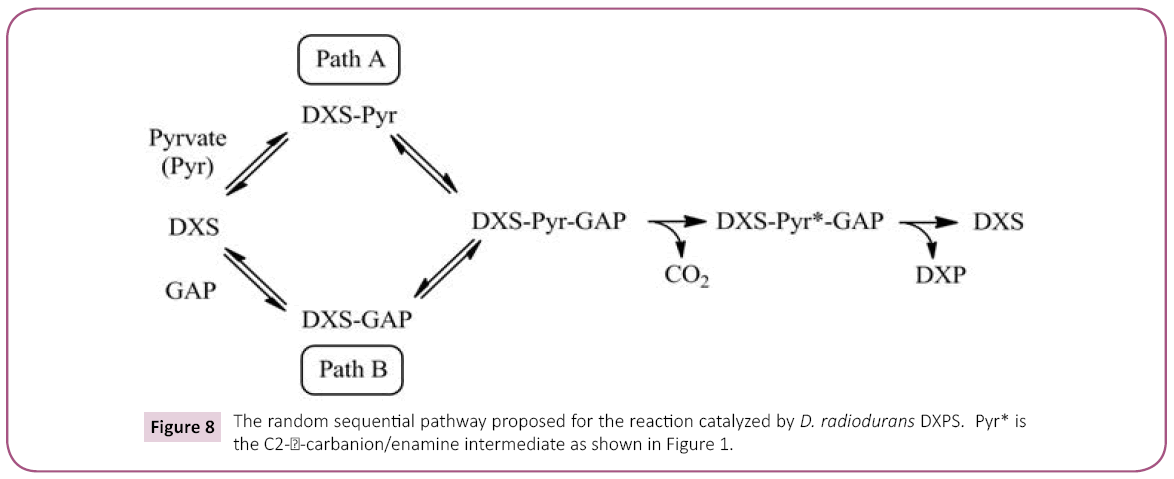
Figure 8: The random sequential pathway proposed for the reaction catalyzed by D. radiodurans DXPS. Pyr* is the C2- -carbanion/enamine intermediate as shown in Figure 1.
-carbanion/enamine intermediate as shown in Figure 1.
Conclusion
We have produced and characterized a series of D. radiodurans DXPS mutants, the selection based upon the sequence alignment of a number of DXPS enzymes and relationships to key amino acids in other TPP-dependent enzymes. Mutant proteins were constructed of these domain I amino acids, His-82, Asn-181, Asn-183, and His-304, and these of domain II, Tyr-395, Arg-423, Asp-430, and His-434. The results of our mutation studies point to differences in the roles for Arg-423, Asp-430, and His-434 in substrate binding and catalysis for D. radiodurans DXPS relative to the equivalent amino acids in transketolase, another TPPdependent enzyme. Since Arg-423, Asp-430, and His-434 are all found within domain II in D. radiodurans DXPS our data point towards broad differences in the role of domain II in substrate binding and catalysis in DXPS relative to transketolase. Another outcome from our mutagenesis work on D. radiodurans DXPS is identification of a potential “back-up” role between His-82 and His-304 in catalysis meaning that one of these His residues can fulfill the catalytic role of the other when the other His is converted to Ala by mutagenesis. We complemented our mutagenesis studies with experiements demonstrating that the kinetic mechanism of D. radiodurans DXPS is, most likely, rapid equilibrium random similar to that reported for E. coli DXPS. DXPS catalyzes the first step in the non-mevalonate pathway of isoprenoid biosynthesis in many pathogens. DXPS and the other enzymes of this pathway represent drug targets as these enzymes have no homology the enzymes of the mevalonate-dependent pathway of isoprenoid biosynthesis in man and other mammals. The work presented herein contributes to our understanding of TPP-dependent catalysis and provides information about the active site architechure and order of substrate binding for D. radiodurans DXPS. These data should aid in the rational design of DXPS inhibitors that to attack pathogens reliant on nonmevalonate pathway for their isoprenoids.
Acknowledgements
This research was supported in part by a seed grant from the Florida Center of Excellence for Biomolecular Identification and Targeted Therapeutics (FCoE-BITT), the University of South Florida (a Research Scholarship grant from the College of Arts and Sciences), the Shirley W. and William L. Griffin Foundation, and National Institutes of Drug Abuse at the National Institutes of Health (R03?DA034323) to D.J.M. and a Graduate Multidisciplinary Scholar (GMS) award from FCoE-BITT to S.H. The other authors, D.R., N.C., D.R.D., W.C.G., T.J.S, J.K.W., and H.L.W., report no declarations of interest.
References
- Friedman M, Byers SO, St. George S (1956) Cholesterol metabolism. Ann Rev Biochem25: 613-640.
- Miller WL, Auchus RJ (2011) The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorder. Endocr Rev 32: 81-151.
- Popják G (1958) Biosynthesis of cholesterol and related substances. Annu Rev Biochem 27: 533-560.
- Cornforth JW (1959) Biosynthesis of fatty acids and cholesterol considered as chemical processes. J Lipid Res 1: 3-28.
- Bucher NLR, Overath P, Lynen F (1960) Beta-hydroxy-beta-methylglutaryl coenzyme A reductase, cleavage and condensing enzymes in relation to cholesterol formation in rat liver. BiochimBiophysActa 40: 491-501.
- Bloch K (1965) The biological synthesis of cholesterol. Science 150: 19-28.
- Gräwert T, Groll M, Rohdich F, Bacher A, Eisenreich W (2011) Biochemistry of the non-mevalonateisoprenoid pathway, Cell Mol Life Sci 68: 3797-3814.
- Hunter WN (2007) The non-mevalonate pathway of isoprenoid precursor biosynthesis. J BiolChem 282: 21573-21577.
- Van der Meer JY, Hirsch AKH (2012) TheNat Prod Rep 29: 721-728.
- Sprenger GA, Schörken U, Wiegert T, Grolle S, De Graaf AA, et al. (1997) Identification of a thiamin-dependent synthase in Escherichia coli required for the formation of the 1-deoxy-D-xylulose 5-phosphate precursor to isoprenoids, thiamin, and pyridoxol. Proc Natl Acad Sci USA 94: 12857-12862.
- Müller IB, Hyde JE, Wrenger C (2010) Vitamin B metabolism in Plasmodium falciparum as a source of drug targets. Trends Parasitol 26: 35-43.
- Begley TP (1996) The biosynthesis and degradation of thiamin (vitamin B1). Nat Prod Rep 13: 177-185.
- Drewke C, Leistner E (2001) Biosynthesis of vitamin B6 and structurally related derivatives. Vitam Horm 61: 121-155.
- Estévez JM, Cantero A, Reindl A, Reichler S, León P (2001) 1-Deoxy-D-xylulose-5-phosphate synthase, a limiting enzyme for plastidic isoprenoid biosynthesis in plants. J Biol Chem 276: 22901-22909.
- Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR (1989) Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 77: 51-59.
- Humnabadkar V, Jha RK, Ghatnekar N, De Sousa SM (2011) A high-throughput screening assay for simultaneous selection of inhibitors of Mycobecterium tuberculosis 1-deoxy-D-xylulose-5-phosphate synthase (Dxs) or 1-deoxy-D-xylulose-5-phosphate reductoisomerase (Dxr). J Biomol Screen 16: 303-312.
- Brammer LA, Meyers CF (2009) Revealing substrate promiscuity of 1-deoxy-D-xylulose 5-phosphate synthase. Org Lett 11: 4748-4751.
- Hawkins CF, Borges A, Perham RN (1989) A common structural motif in thiamin pyrophosphate-binding enzymes. FEBS Lett 255: 77-82.
- Xiang S, Usunow G, Lange G, Busch M, Tong L (2007) Crystal structure of 1-deoxy-D-xylulose 5-phosphate synthase, a crucial enzyme for isoprenoids biosynthesis. J BiolChem 282: 2676-2682.
- Xiang S, Usunow G, Lange G, Busch M, Tong L (2013) 1-Deoxy-D-xylulose 5-phosphate synthase (DXS), a crucial enzyme for isoprenoids biosynthesis. In: Bach TJ, Rohmer M, (eds)Isoprenoidsynthesis in plants and microorganisms: New concepts and experimental approaches. Springer Science + Business Media, New York, USA. 13: 17-28.
- Arjunan P, Nemera N, Brunskill A, Chandrasekhar K, Sax M, et al. (2002) Structure of pyruvate dehydrogenase multienzyme complex E1 component from Escherichia coli at 1.85 Å resolution. Biochemistry 41: 5213-5221.
- Nikkola M, Lindqvist Y, Schneider G (1994) Refined structure of transketolase from Saccharomyces cerevisiae at 2.0 Å resolution. J MolBiol 238: 387-404.
- Pang SS, Duggleby RG, Guddat LW (2002) Crystal structure of yeast acetohydroxyacid synthase: A target for herbicidal inhibitors. J MolBiol 317: 249-262.
- Takahashi K, Tagami U, Shimba N, Kashiwagi T, Ishikawa K, et al. (2010) Crystal structure of Bifidobacteriumlongumphosphoketolase; Key enzyme for glucose metabolism in Bifidobacterium. FEBS Lett 584: 3855-3861.
- BreslowR (1958) On the mechanism of thiamine. IV. Evidence from studies on model systems. J Am ChemSoc 80: 3719-3726.
- Guo F, Zhang D, Kahyaoglu A, Farid RS, Jordan F (1998) Is a hydrophobic amino acid required to maintain the reactive V conformation of thiamin at the active center of thiamin diphosphate-requiring enzymes? Experimental and computational studies of isoleucine 415 of yeast pyruvate decarboxylase. Biochemistry 37: 13379-23391.
- Duggleby RG (2006) Domain relationships in thiamine diphosphate-dependent enzymes. AccChem Res 39: 550-557.
- Wikner C, Nilsson U, Meshalkina L, Udekwu C, Lindqvist Y, et al. (1997) Identification of catalytically important residues in yeast transketolase. Biochemistry 36: 15643-15649.
- Fiedler E, Thorell S, Sandalova T, Golbik R, König S, et al. (2002) Snapshot of a key intermediate in enzymatic thiamin catalysis: crystal structure of the alpha-carbanion of (alpha,beta-dihydroxyethyl)-thiamin diphosphate in the active site of transketolase from Saccharomyces cerevisiae. ProcNatlAcadSci USA 99: 591-595.
- Nilsson U, Meshalkina L, Lindqvist Y, Schneider G (1997) Examination of substrate binding in thiamin diphosphate-dependent transketolase by protein crystallography and site-directed mutagenesis. J BiolChem 272: 1864-1869.
- Schneider G, Lindqvist Y (1998) Crystallography and mutagenesis of transketolase: mechanistic implications for enzymatic thiamin catalysis. BiochimBiophysActa 1385: 387-398.
- Singleton CK, Wang JJL, Shan L, Martin PR (1996) Conserved residues are functionally distinct within transketolases of different species. Biochemistry 35: 15865-15869.
- Yevenes A, Frey PA (2008) Cloning, expression, purification, cofactor requirements, and steady state kinetics of phosphoketolase-2 from Lactobacillus plantarum. BioorgChem 36: 121-127.
- Liu S, Gong X, Yan X, Peng T, Baker JC, et al. (2001) Reaction mechanism for mammalian pyruvate dehydrogenase using natural lipoyl domain substrates. Arch BiochemBiophys 386: 123-135.
- Frank RAW, Titman CM, Pratap JV, Luisi BF, Perham RN (2004) A molecular switch and proton wire synchronize the active sites in thiamine enzymes. Science 306: 872-876.
- Eubanks LM, Poulter CD (2003) Rhodobactercapsulatus 1-deoxy-D-xylulose 5-phosphate synthase: steady-state kinetics and substrate binding. Biochemistry 42: 1140-1149.
- Brammer LA, Smith JM, Wade H, Meyers CF (2011) 1-Deoxy-D-xylulose 5-phosphate synthase catalyzes a novel random sequential mechanism. J BiolChem 286: 36522-36531.
- Patel H, Nemeria NS, Brammer LA, Freel-Meyers CL (2012) Observation of thiamin-bound intermediates and microscopic rate constants for their inter-conversion on 1-dexoy-D-xylulose 5-phosphate synthase: 600-fold rate acceleration of pyruvate decarboxylation by D-glyceraldehyde-3-phosphate. J Am ChemSoc 134: 18374-18379.
- Brammer-Basta LA, Patel H, Kakalis L, Jordan F, Freel-Meyers CL (2014) Defining critical residues for substrate binding to 1-deoxy-D-xylulose 5-phosphate synthase – active site substitutions stabilize the pre-decarboxylation intermediate C2-α-lactylthiamindiphosphate. FEBS J 281: 2820-2837.
- Cook P, Cleland WW (2007) Enzyme kinetics and mechanism. Garland Science, London, UK.
- Prasanna KV, West AH, Cook PF (2011) Kinetic and chemical mechanisms of homocitrate synthase from Themusthermophilus. J BiolChem 286: 29428-29439.

