Keywords
Vitamin K; Protein; VKOR
Introduction
The vitamin K epoxide reductase (VKOR) activity has been described since the early 70s’ [1]. This VKOR activity allows the reduction of vitamin K epoxide in order to regenerate vitamin K quinone. This activity is a key step of the vitamin cycle and is the target of vitamin K antagonist (VKA). The VKORC1 gene has only been identified in 2004 [2,3]. This gene encodes for a 163 amino acid protein with, probably, four transmembrane domains and a luminal loop. This protein contains also a C132XXC135 redox motif, as previously hypothesized by Silverman [4]. This motif is located in the fourth transmembrane domain.
VKA actively inhibit VKORC1 in the liver and thus limit the gamma-carboxylation of hepatic VKD proteins resulting in an intense anticoagulant effect. VKA are extensively used worldwide for prevention and treatment of thromboembolic disorders. Many missenses mutations have been detected in VKORC1 gene in humans and rats and are sometimes associated to a VKA resistance phenotype [5-7].
Until 2011, VKORC1 was considered as the only protein able to catalyze the VKOR activity. Nevertheless, Westhofen et al. [8] described that a paralogous enzyme of VKORC1, the VKORC1L1, could also reduce vitamin K>0 to vitamin KH2 when expressed in HEK293T cells. Resulting from a gene duplication of an early common VKOR ancestor, there is 50% of homology between this protein and VKORC1 [9]. The VKORC1L1 gene encodes for a 176 amino acids protein with almost the same structure than VKORC1. However, this VKOR activity was described with a low enzymatic efficiency and then they propose that VKORC1L1 is responsible for driving the vitamin K mediated intracellular antioxidation pathway critical to cell survival by generating vitamin KH2 [8].
In another hand, Hammed et al. [10] emphasized two main differences between VKORC1L1 and VKORC1. VKORC1L1- catalyzed VKOR activity is 30-fold more resistant to warfarin than that catalyzed by VKORC1. The tissue distributions of the corresponding mRNA are quite different. Liver expression of Vkorc1L1 mRNA is very low in comparison to Vkorc1. On the contrary, in other tissues such as lung or testis, the expression of these two genes is similar. These data argue for the fact that VKORC1L1 does not interfere with the coagulation process which is supported by the liver. On the contrary, these data argue for the fact that VKORC1L1, being resistant to VKA, could protect extrahepatic tissues against the effects of VKAs. These results provide an interesting explanation for the low or even the absence of clinical consequences on the gamma carboxylation of vitamin K-dependent proteins (VKDP) produced by extra-hepatic tissues such as matrix Gla protein (MGP) [11], osteocalcin (OC) [12] or Gas 6 [13]. Indeed, the absence of gamma carboxylation of MGP should create ectopic mineralization [14] and the absence of gamma carboxylation of osteocalcin should have consequences on energetic metabolism [15,16].
Unlike VKORC1 proteins, which have been subjected to extensive investigation, our understanding of the mechanism of action of VKORC1L1 is limited. As the contrast between the susceptibility to VKAs of VKORC1 and VKORC1L1 appears to be an important determinant for the biological function of the VKORC1L1 protein, the objective of this paper is to analyze the structural basis of the natural resistance of VKORC1L1 to VKAs in the absence of 3D-structures of VKORC1L1 proteins. To highlight important residues for VKORC1L1 function, sequences alignments were first performed between VKORC1 proteins and VKORC1L1 proteins. Finally, using site directed mutagenesis, we analyzed the functional implications of these highlighted amino acid residues. Our results suggest the central function of Glu-130(+7) amino acid residue in the natural resistance to VKAs of VKORC1L1 compared with VKORC1.
Methods
Plasmid constructions
Human VKORC1 and VKORC1L1 coding sequences fused with a c-myc tag via a flexible (GGS)3 in its 3’-extremity was optimized for heterologous expression in yeast and synthesized by GenScript (Piscataway, NJ, USA). Synthesized nucleotide sequences included EcoRI and XbaI restriction sites at their 5’- and 3’-extremities, respectively. These nucleotide sequences were subcloned into pPICZ-B (Invitrogen, Cergy Pontoise, France) and sequenced on both strands. Construction of amino acid substituted mutants of VKORC1 or VKORC1L1 was carried out using pPICZ-VKORC1 or pPICZ-VKORC1L1 as template, respectively, with the QuikChange site-directed mutagenesis kit (Stratagene). Mutants were systematically checked by sequencing, and the various mutants were individually expressed in P. pastoris.
Heterologous expression in Pichia pastoris
Heterologous expressions of VKORC1 and VKORC1L1 proteins were performed in Pichia pastoris as described previously [5,17]. pPICZ-VKORC1 or VKORC1L1 vectors were individually transformed into the P. pastoris SMD1168 yeast strain using the P. pastoris Easy Comp Transformation kit (Invitrogen). Transformants were selected on YPD plates (1% (w/v) yeast extract, 2% (w/v) peptone, 2% (w/v) dextrose) containing 100 μg/ ml Zeocin (Invitrogen). The cells were grown in BMGY medium (1% (w/v) yeast extract, 2% (w/v) peptone, 100 mM potassium phosphate, pH 6.0, 1.34% (w/v) yeast nitrogen base, and 1% (v/v) glycerol). Expression was induced by methanol (1%, v/v) for 48 h at 30°C in a rotary shaker (200 rpm). Yeast cells were collected by centrifugation (3000 g-10 min) and immediately frozen at âÂâ€Â€20°C.
Subcellular fractionation of recombinant yeast cells
Yeast microsomes were prepared from thawed yeast cells by differential centrifugation. Briefly, yeast cells were resuspended in 50 mM Phosphate Buffer (pH 7.4) containing 1.15% (w/v) of KCl. Yeast cells were broken with Zircon beads using Dispermat® LC30 (VMA-GETZMANN, germany) (15 min-3500 rpm) a continuously at 4°C and further submitted to differential centrifugation. The 100,000 g pellet corresponding to the membrane fraction was resuspended by Potter homogenization in HEPES glycerol buffer (50 mM Hepes, 20% glycerol, pH 7.4). Protein concentrations were evaluated by the method of Bradford using bovin serum albumin as a standard. Microsomes were frozen at –80°C and used for kinetic analysis.
Immunoblot analysis
Expression level quantification of VKORC1 and VKORC1L1 proteins in microsomal fractions were determined by western blotting as described previously [6]. Microsomal proteins were separated on 12% SDS-polyacrylamide gel electrophoresis, transferred onto Immobilon-P membranes and probed with anti-c-myc antibodies (Invitrogen, Cergy Pontoise, France). The resulting immunocomplexes were visualized using alkaline phosphataseconjugated anti-mouse immunoglobulins as secondary antibodies and a BCIP/NBT solution. Quantification of the stained bands was performed by densitometry using the Scion Image software. The relative intensity (RI) of the signal was correlated to the quantity of microsomal proteins. The relation was linear from 0 to 50 μg for microsomal proteins.
To evaluate the expression level of VKORC1 and VKORC1L1 proteins, the expression of wild type human VKORC1 was designated as the basal expression. For the quantification of wild type or mutant VKORC1 or VKORC1L1, the same unique pool of yeast microsomes containing human VKORC1 was used. Therefore, its expression factor was by definition 1. The expression level of VKORC1 or VKORC1L1 proteins was evaluated by comparison to the expression of human VKORC1. For this purpose, various amounts (from 0 to 20 μg) of microsomal proteins containing human VKORC1 and various amounts (depending on the expression level) of microsomal proteins containing VKORC1 or VKORC1L1 proteins were analyzed on the same western blot. Two linear relations (RI = a × quantity of microsomes loaded) were obtained, the first one for microsomes containing human VKORC1 (characterized by a specific slope ahVKORC1), the second one for microsomes containing VKORC1 or VKORC-1L1 proteins (characterized by a slope atarget). Ratio atarget/ahVKORC1 allowed us to determine the expression factor characterizing the expression level of VKORC1 or VKORC1L1 proteins in the microsomal fraction compared with the expression level of the human VKORC1.
Vitamin K epoxide reductase activity (VKOR) assays and kinetics
Microsomal vitamin K epoxide reductase (VKOR) activity was assayed as described previously [5,17]. Briefly, standard reactions were performed in 200 mM Hepes buffer (pH 7.4) containing 150 mM KCl, 1 mM dithiothreitol, 0.25 to 2 g.L-1 of total proteins containing membrane VKORC1. The reaction was started by the addition of vitamin K1>0 solution in 1% Triton X-100 and incubated at 37°C for 30 min. In these conditions, the reaction was linear according to the time of incubation and the quantity of incubated proteins. After incubation at 37°C for 30 min, the reaction was stopped by adding of 4 mL of iced 1:1 isopropanol/ hexane solution. After centrifugation at 5000 g for 10 min, the hexane layer was removed and dried under nitrogen. The dry residue was immediately dissolved in 0.2 mL of isopropanol and reaction product was analyzed by liquid chromatography-mass spectrometry.
The LC-APCI/MS/MS used was a 1100 Series LC/MSD ion Trap VL with an Atmospheric Pressure Chemical Ionisation (APCI) interface and a LCMS Chemstation software from Agilent Technologies (Palo Alto, CA, USA). Chromatographic separation was performed using a SunFire reverse phase C8 column (4.6 mm × 150 mm, 5 μm, Waters, Milford, MA, USA) with a mobile phase of methanol, 0.1% acetic acid (95:5) in isochratic conditions. The column temperature was 45°C. The flow rate in the LC column was 1 ml/min. The injection volume was 20μl. The temperature of the autosampler tray was set to 5°C and the samples were protected from the daylight. Detection was by MS/MS with APCI source in positive mode. Nebulizer pressure was set to 60psi, dry gas temperature to 350°C, dry gas flow to 5 L/min, and vaporizer temperature to 400°C. Capillary voltage was set to 4000V, corona needle to 4000 nA, and CID to 1V. Collision gas in the trap was helium with a pressure of 0.6*10-5 mbar. Identification criteria for vitamin K1 are the retention time (tr= 4.1 min) and the product ion 451 ≥ 187 (m/z(+)=187). Identification criteria for vitamin K1>0 are the retention time (tr=3.4 min) and the product ion 467 ≥ 307 (m/z(+)=307). Linearity and accuracy were tested from 25 to 2000 ng/ml (n=20). The response was linear throughout the concentration range tested with a coefficient of correlation (r2) above 0.99. Accuracy was between 80 and 120% of the theoretical concentrations.
Km, Vmax and Ki values were obtained from at least three separate experiments performed on two different batches of protein. The estimation of Km and Vmax values was achieved by the incubation of at least 9 different concentrations of vit K>O (from 0.003 to 0.2 mM) to the standard reaction. Incubations were performed in duplicate. Data were fitted by nonlinear regression to the Michaelis-Menten model using the R-fit program. In order to evaluate the inhibiting effect of warfarin on VKOR activity, Ki were determined after addition of various concentrations of anticoagulant to the standard reaction in the presence of increasing amounts of vit K>O (from 0.003 to 0.2 mM) using anticoagulant concentrations from about 0.05 to 20×Ki. Data were fitted by non-linear regression to the non-competitive inhibition model v= (Vmax/(1+(I/Ki)))*(S/(Km+S)) using the R-fit program.
Results
Comparative inhibition of VKOR activity catalysed by VKORC1 or VKORC1L1
The susceptibility of wild type VKORC1L1 to various VKAs used either in human medicine or as rodenticide was evaluated comparatively to that of wild type VKORC1. Results are presented in Table 1. While VKORC1 was inhibited by all the VKAs used in this study with Ki lower than 0.5 μM (except for warfarin and phenpromoumon), VKORC1L1 was particularly resistant to some VKAs with Ki systematically higher than 0.5 μM. More exactly, VKORC1L1 was roughly 30-fold more resistant to warfarin, a 4-hydroxycoumarin-derivative and the most VKA used worldwide, than VKORC1 (52 μM vs 1.7 μM). In the same way, fluindione, an indanedione derivative largely used in France was completely ineffective to inhibit VKORC1L1 with a Ki 80-fold higher than that obtained for VKORC1 (21.3 μM vs 0.25 μM). Acenocoumarol and phenprocoumon were more effective to inhibit VKORC1L1 than warfarin and fluindione, but Ki obtained (i.e., 2.7 and 5.4 μM, respectively) were still largely higher than those obtained for VKORC1 (i.e., 0.3 and 0.7 μM, respectively). VKORC1L1 was also resistant to tecarfarin, a new VKA developed to avoid the CYP- 450-dependent metabolism pathway [16]. Ki towards tecarfarin obtained with VKORC1L1 was 30-fold higher than that obtained with VKORC1 (2.4 vs 0.08 μM). The most potent VKAs, such as difenacoum, brodifacoum and difethialone, which possess very low Ki towards VKORC1, were also assayed in this study. These VKAs are used as rodenticide due to their good efficiency even on resistant strains of rats. Even towards these very effective VKAs, VKORC1L1 was approximately 7-fold more resistant than VKORC1, (Difenacoum 0.7 μM vs 0.1 μM, Brodifacoum 0.6 μM vs 0.15 μM, Difethialone 0.9 μM vs 0.2 μM).
| Molecule |
Chemical structure |
Ki hVKORC1
(µM) |
Ki hVKORC1L1
(µM) |
Ki hVKORC1L1 / KihVKORC1 |
| Warfarin |
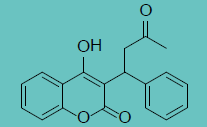 |
1.65 ± 0.79 |
52.0 ± 3.0 |
32 |
| Acenocoumarol |
 |
0.33 ± 0.18 |
2.69 ± 0.8 |
8 |
| Phenprocoumon |
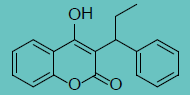 |
0.72 ± 0.31 |
5.36 ± 1.6 |
7 |
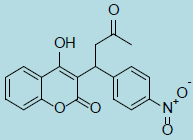
| Fluindione |
 |
0.25 ± 0.14 |
21.3 ± 1.3 |
85 |
| Tecarfarine |
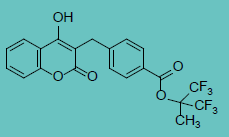 |
0.08 ± 0.01 |
2.40 ± 0.40 |
30 |
| Difenacoum |
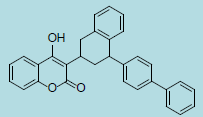 |
0.12 ± 0.03 |
0.71 ± 0.09 |
6 |
| Brodifacoum |
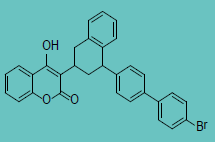 |
0.15 ± 0.02 |
0.57 ± 0.08 |
4 |
| Difethialone |
 |
0.18 ± 0.03 |
0.87 ± 0.16 |
5 |
Table 1 Ki values of human VKORC1 and human VKORC1L1 proteins towards various VKA. Inhibition parameters were assessed using anticoagulant concentrations from about 0.05 to 20×Ki.
VKORC1 and VKORC1L1 alignment
To gain insight into potential functional domains or amino acid residues of VKORC1L1 involved in VKOR activity and/or resistance to VKAs in the absence of 3D-structure of VKORC1L1 proteins, we performed a multiple alignment of 20 selected VKORC1L1 sequences plus 26 VKORC1 sequences, taken as an outgroup (Figures 1 and 2). VKORC1L1 proteins contain 176 amino acid residues, while VKORC1 proteins contain 163 amino acid residues. The sizes of the N-terminal and the C-terminal domains are longer in the VKORC1L1 group compared with the 26 VKORC1 proteins. The sequence comparison highlighted i) a very strong identity between VKORC1L1 sequences (between 85 % and 100 % according to the species considered) and ii) a limited degree of identity between VKORC1L1 sequences and VKORC1 sequences (~50% of identity) with two remarkable conserved peptide domains with an identity higher than 75%. More precisely, 47 amino acid positions are identical, both in the VKORC1 outgroup and in the VKORC1L1 family. Among these conserved positions, 9 are located in the first conserved motif and 17 are located in the second conserved motif; the others are distributed all along the alignment.
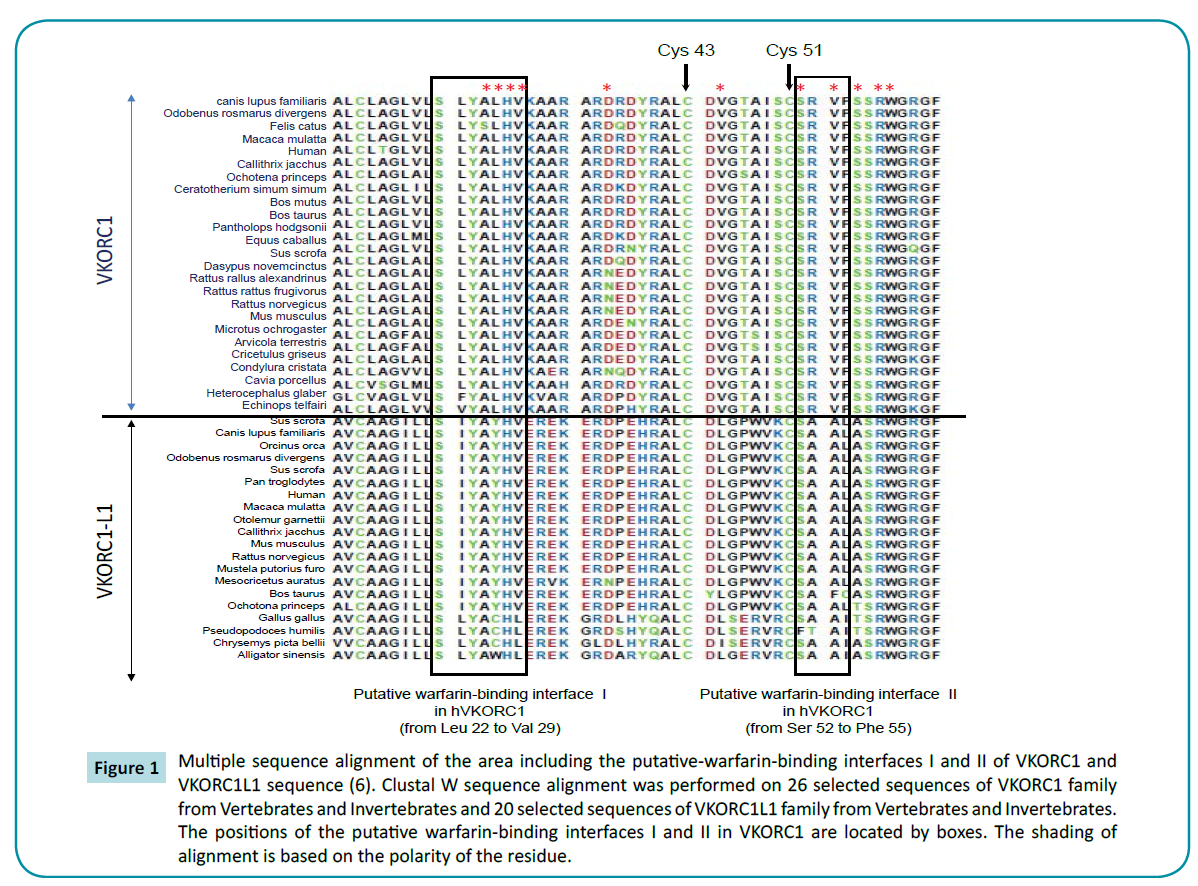
Figure 1 Multiple sequence alignment of the area including the putative-warfarin-binding interfaces I and II of VKORC1 and VKORC1L1 sequence (6). Clustal W sequence alignment was performed on 26 selected sequences of VKORC1 family from Vertebrates and Invertebrates and 20 selected sequences of VKORC1L1 family from Vertebrates and Invertebrates. The positions of the putative warfarin-binding interfaces I and II in VKORC1 are located by boxes. The shading of alignment is based on the polarity of the residue.
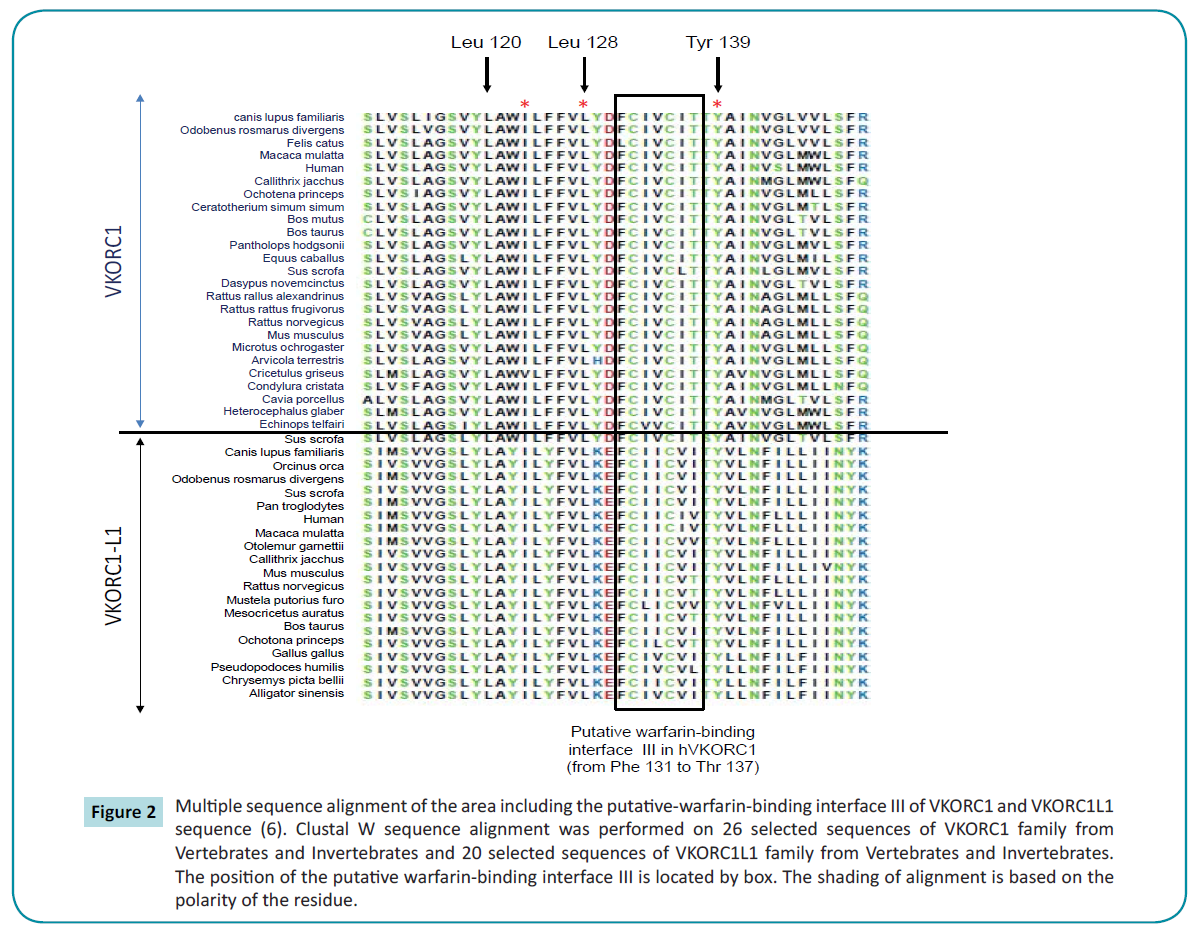
Figure 2 Multiple sequence alignment of the area including the putative-warfarin-binding interface III of VKORC1 and VKORC1L1 sequence (6). Clustal W sequence alignment was performed on 26 selected sequences of VKORC1 family from Vertebrates and Invertebrates and 20 selected sequences of VKORC1L1 family from Vertebrates and Invertebrates. The position of the putative warfarin-binding interface III is located by box. The shading of alignment is based on the polarity of the residue.
The first conserved domain from amino acid 64 to amino acid 72 of VKORC1L1 sequences (i.e., the “SRWGRGFGL” motif) is strictly conserved between VKORC1L1 and VKORC1 sequences (Figure 1). Nevertheless, none of these residues were described to be of importance in VKORC1. Close to the first domain are found the Cys-43 and Cys-51 strictly conserved in VKORC1 proteins and proposed as important residues in VKORC1 proteins. These Cys are also strictly conserved in VKORC1L1 proteins in position 50 and 58, respectively (Figure 1).
The second conserved domain between VKORC1 and VKORC1L1 proteins contain amino acid 123 to amino acid 146 in VKORC1L1 (i.e., corresponding to the residues 116 to 139 in VKORC1). This domain is highly conserved with 75% of identity between VKORC1L1 sequences and VKORC1 sequences (Figure 2). This second domain contains the “CXXC” motif proposed as catalytic site in the VKORC1 proteins. This “CXXC” motif is also present in VKORC1L1 proteins from amino acid position 139 to 142 (Figure 2). Close to this domain are found the main amino acid positions of VKORC1 leading to severe resistance to VKA (i.e., amino acid position 120, 128 and 139). Leu-120, Leu-128 and Tyr-139 strictly conserved between VKORC1 proteins are also strictly conserved between VKORC1L1 in position 127, 135 and 146, respectively (Figure 2). On the other hand, because the hypothetical catalytic mechanism proposed by Silverman [3] for VKORC1 implies the involvement of nucleophilic amino acid residues close to the catalytic site, the conservation of nucleophilic amino acid residues between VKORC1 and VKORC1L1 proteins inside the second conserved domain was analyzed. In VKORC1, three nucleophilic residues close to the catalytic site are found in position 129 (i.e., Tyr), 130 (i.e., Asp) and 139 (i.e., Tyr). In VKORC1L1 proteins, the first nucleophilic amino acid is not conserved and is replaced by a basic amino acid found in position 136 (i.e., Lys) but totally conserved between VKORC1L1 proteins. The two other nucleophilic residues close to the catalytic site conserved between VKORC1 are also conserved in VKORC1L1 proteins and correspond to Tyr-146 and Glu-137.
The presence of 7 additional amino acids in the first 20 amino acids of VKORC1L1 leads to a gap of the numbering of amino acid residues between VKORC1 and VKORC1L1 sequences. To facilitate the understanding of the results, we decide to number amino acids of VKORC1L1 by the number of the corresponding amino acid in VKORC1 and to add to this number (+7) in index.
Analysis of the importance of the “CXXC” motif in the function of VKORC1L1
To address the functional contribution of the “CXXC” motif in the vitamin K epoxide reductase activity catalyzed by VKORC1L1, we conducted a systematic site-directed mutagenesis approach. Individual cysteines were subjected to non-conservative substitutions to generate 2 mutants VKORC1L1-C132(+7)A and VKORC1L1-C135(+7)A. Wild-type and mutant VKORC1L1 enzymes were expressed at a similar level in Pichia pastoris (data not shown) and tested for in vitro vitamin K epoxide reductase activity using K1>0 as substrate. Activity of the recombinant enzymes determined in the presence of 200 μM of K1>0 was normalized to the amount of expressed protein, and values are presented in Figure 3. The recombinant wild type VKORC1L1 exhibited high activity up to 20 pmol·min−1·mg protein, although it was undetectable in mock-transformed yeasts. On the other hand, replacement of each cysteine of the “CXXC” motif completely abolished the in vitro enzyme activity (i.e. less than 2% of the recombinant VKORC1L1 activity), emphasizing the prevalent function of the “CXXC” motif for the vitamin K epoxide reductase activity of VKORC1L1.
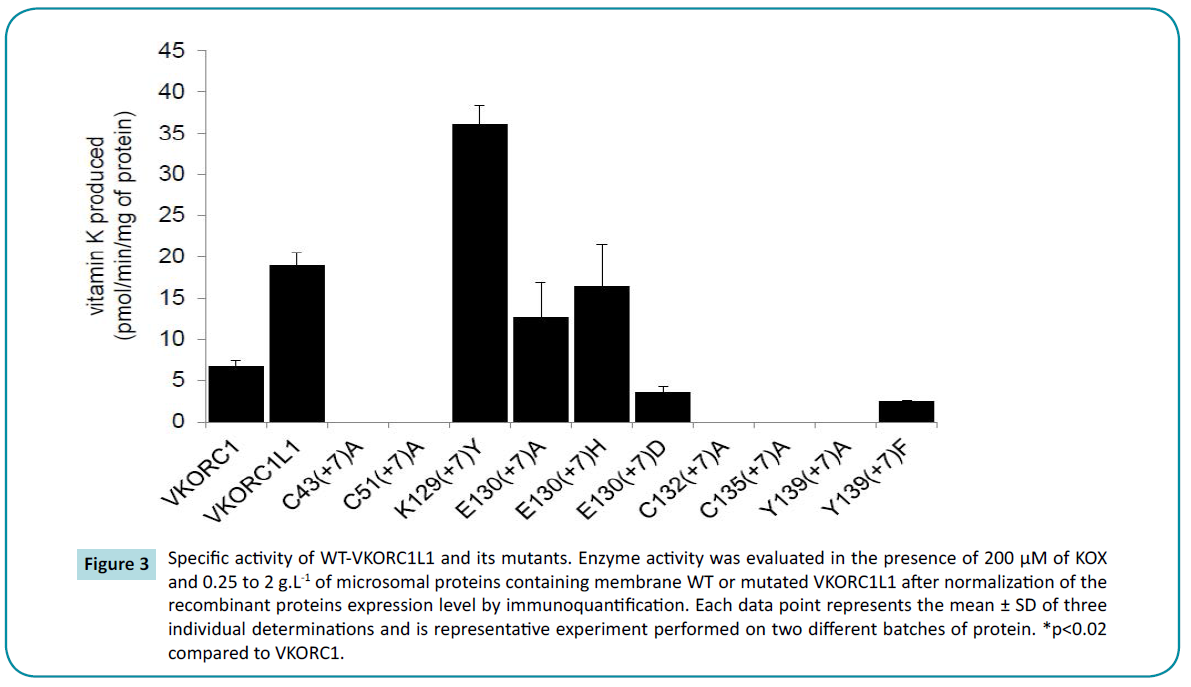
Figure 3 Specific activity of WT-VKORC1L1 and its mutants. Enzyme activity was evaluated in the presence of 200 μM of KOX and 0.25 to 2 g.L-1 of microsomal proteins containing membrane WT or mutated VKORC1L1 after normalization of the recombinant proteins expression level by immunoquantification. Each data point represents the mean ± SD of three individual determinations and is representative experiment performed on two different batches of protein. *p<0.02 compared to VKORC1.
Analysis of the importance of Cys-43(+7) and Cys- 51(+7) in the function of VKORC1L1
To further investigate the importance of conserved Cys-43(+7) and Cys-51(+7) in the VKOR activity catalyzed by VKORC1L1, we analyzed the catalytic effects of non-conservative mutation of these residues into Ala. Upon expression, VKORC1L1-C43(+7) A and VKORC1L1-C51(+7)A mutants were expressed at similar or higher levels than that of the wild-type enzyme (data not shown). Interestingly, replacement of Cys-43(+7) or Cys-51(+7) of VKORC1L1 by Ala abolished the in vitro VKOR activity (i.e. less than 2% of the recombinant VKORC1L1 activity) in the presence of saturating concentration of K1>0, suggesting the major role of both Cys in the function of VKORC1L1.
Analysis of the importance of the nucleophilic amino acid residues surrounding the “CXXC” motif of VKORC1L1
In the next series of experiments, we explored the specific function of Lys-129(+7), Glu-130(+7) and Tyr-139(+7)) amino acid residues surrounding the “CXXC” motif. To address the importance of these invariant amino acids in VKOR activity, a series of mutants exhibiting non-conservative and/or conservative substitutions at each position were engineered to generate 6 mutants VKORC1L1-K129(+7)Y, VKORC1L1-E130(+7)A, -E130(+7)H, -E130(+7) D, VKORC1L1-Y139(+7)A and -Y139(+7)F. The recombinant proteins were produced in yeast P. pastoris. Immunoblot analysis indicated that these mutants were all produced and their expression factor was comprised between 0.3 to 2 compared with the expression of recombinant wild type VKORC1 protein used as a standard (data not shown).
To evaluate the consequences of the mutations on the enzyme function, we analyzed the activity of mutated VKORC1L1 proteins (Figure 3). VKORC1L1-K129(+7)Y exhibited up to 200% vitamin K epoxide reductase activity in the presence of 200 μM of K1>0 compared with wild-type VKORC1L1 enzyme ; VKORC1L1-E130(+7) A and VKORC1L1-E130(+7)H exhibited up to 67 and 86% vitamin K epoxide reductase activity. The non-conservative mutation of Tyr- 139(+7) in Ala abolished the VKOR activity suggesting the important role of this residue in VKOR activity (Figure 3). On the contrary, the replacement of Tyr-139(+7) by Phe, another aromatic amino acid residue led to a mutant that exhibited up to 14% vitamin K epoxide reductase activity, indicating that Phe and Tyr residues are to some extent interchangeable at this position. Kinetic parameters and susceptibility to VKA were thus determined for active mutated VKORC1L1 proteins and are presented in Tables 2 and 3, respectively.
| Protein |
Km
(µM) |
Vmax
(pmol.min-1.mg-1) |
Vmax/Km
(nL.min-1.mg-1) |
| hVKORC1 |
21.5 ± 4.2 |
7.1 ± 0.5 |
330 |
| hVKORC1L1 |
24.1 ± 3.0 |
20.7 ± 0.9 |
859 |
| K129(+7)Y |
22.2 ± 11.8 |
39.2 ± 5.1 |
1766 |
| E130(+7)A |
29.8 ± 8.9 |
13.1 ± 4.4 |
440 |
| E130(+7)H |
31.1 ± 8.3 |
19.0 ± 5.1 |
611 |
| E130(+7)D |
19.1 ± 5.7 |
3.8 ± 0.7 |
199 |
| Y139(+7)A |
_ |
_ |
_ |
| Y139(+7)F |
25.1 ± 1.2 |
2.7 ± 0.1 |
108 |
Table 2 Apparent kinetic parameters towards K>0 of human VKORC1, VKORC1L1 and mutated VKORC1L1 proteins. Vmax values determined at saturating concentration of K>O were evaluated after normalization of the recombinant proteins expression level by immunoquantification. Each data point represents the mean ± SD of three individual determinations. * p<0.02 compared to WT.
| Protein |
Ki
(µM) |
| hVKORC1 |
1.65 ± 0.79 |
| hVKORC1L1 |
52.0 ± 0.79 |
| K129(+7)Y |
12.3 ± 1.9 |
| E130(+7)A |
3.2 ±1.1 |
| E130(+7)H |
3.6 ± 1.1 |
| E130(+7)D |
3.2 ± 0.8 |
| Y139(+7)A |
_ |
| Y139(+7)F |
>300 |
Table 3 Ki values of human VKORC1, VKORC1L1 and mutated VKORC1L1 proteins towards warfarin. Inhibition parameters were assessed using anticoagulant concentrations from about 0.05 to 20×Ki.
Discussion
Even if VKORC1L1 was initially proposed to be responsible for driving the vitamin K mediated intracellular antioxidation pathway [8], its crucial role in the gammacarboxylation of extrahepatic VKDP is becoming increasingly obvious. In the absence of VKA treatment, involvement of VKORC1L1 in the gammacarboxylation of extrahepatic VKDP is at least as important as VKORC1. On the other hand, during long-term anticoagulation therapy with warfarin, VKORC1L1, which is 30-fold more resistant to warfarin than VKORC1 [10], is certainly the only enzyme supporting the VKOR activity in extrahepatic tissues and thus, the gammacarboxylation of extrahepatic VKDPs. Warfarin was the first molecule developed as VKA. Since then, numerous vitamin K-antagonists molecules were developed. While these molecules are all much more potent to inhibit VKORC1 than warfarin, their ability to inhibit VKORC1L1 is still unknown. This question is of major interest if we consider that some of these molecules are also used in human medicine during long-term anticoagulation and that their use could naturally inhibit the coagulation process, but also other extrahepatic VKDP-dependent physiological processes not inhibited by the use of warfarin.
To address this question, recombinant VKORC1L1 was expressed in P. pastoris and VKOR activity was characterized by a DTT-driven VKOR assay, as previously described [5,10]. This experimental approach is a direct approach based on the measurement of the VKORC1 reaction product, the vitamin K. The results obtained by this method, and particularly the resistance factors to VKAs obtained by this method, were successfully confronted to results obtained by ex vivo and in vivo assays [17,18-20]. This experimental approach was recently challenged by other indirect approaches designed as cell-based VKOR assays [6,7,21] based on the measurement of the gamma-carboxylation of overexpressed clotting factors by the constitutive GGCX in the presence of vitamin K hydroquinone produced by the overexpressed VKORC1. Since the results obtained by the different developed cell-based VKOR assays are conflicting between them, we chose intentionally to use the DTT-driven VKOR approach, as previously used [10] to characterize the VKOR activity of VKORC1L1.
Seven molecules, known to inhibit VKORC1, were tested in this study as inhibitor of VKORC1L1. These molecules are either 4-hydroxycoumarin or 4-hydroxy-1-thiocoumarin or indanedione derivatives. Whatever the structure of the molecule is, Ki obtained for VKORC1L1 systematically increases as compared to human VKORC1, as it was already observed with warfarin [10]. Hydrophobicity and length of lateral chain of the molecules seems to favor the interaction of anticoagulant with VKORC1L1 (Table 1) and (Figure 4), as previously described for VKORC1 [17,19]. Contrary to human VKORC1, 4-hydroxycoumarin structures seems to interact with VKORC1L1 more efficiently than indanedione structures. Among these molecules, four are used (i.e., acenocoumarol, phenprocoumon, fluindione) [22] or developed to be used (i.e., tecarfarin) [18,23] in human medicine and three are used as rodenticides (i.e., difenacoum, brodifacoum, difethialone) [24]. All these molecules are able to inhibit human VKORC1L1 in a non-competitive manner, as observed for human VKORC1 [5]. Nevertheless, all the molecules used in human medicine (i.e., acenocoumarol, phenprocoumon, fluindione, tecarfarin) are very bad inhibitors of VKORC1L1 (with Ki higher than 2 μM) while they are able to efficiently inhibit human VKORC1 (i.e., with Ki<0.7 μM). It is thus likely that the use of these molecules during long-term anticoagulation therapy allow VKORC1L1 to still support the VKOR activity in extrahepatic tissues, as previously described for warfarin [10]. The other molecules (i.e., difenacoum, brodifacoum, difethialone) with Ki less than 1 μM are probably able to inhibit VKORC1L1 in extrahepatic tissues and, thus inhibit or at least decrease the gammacarboxylation of extrahepatic VKDP. The use of some of these molecules during long-term anticoagulation could lead to serious unwanted side effects due to the undercarboxylation of extrahepatic VKDP such as OC [12], MGP [11], Gas 6 [13]. Fortunately, these molecules are never used in human medicine and are exclusively used for the pest control management.
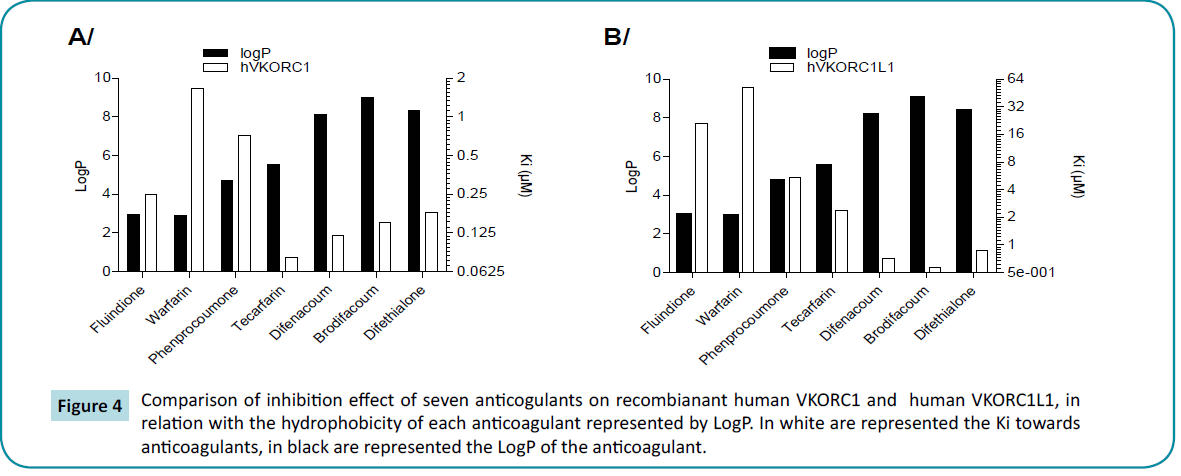
Figure 4 Comparison of inhibition effect of seven anticogulants on recombianant human VKORC1 and human VKORC1L1, in relation with the hydrophobicity of each anticoagulant represented by LogP. In white are represented the Ki towards anticoagulants, in black are represented the LogP of the anticoagulant.
To pinpoint potential amino acid residues of VKORC1L1 involved in catalysis and/or substrate recognition or responsible for the natural resistance to VKAs of VKORC1L1 in the absence of 3D-structure of VKORC1L1 proteins, we carried out a multiple sequences alignment analysis between VKORC1 and VKORC1L1 proteins. This study allowed us to highlight 1) conserved amino acid residues between VKORC1 and VKORC1L1 potentially involved in catalysis (i.e., Cys-43(+7), Cys-51(+7), Asp/Glu-130(+7), Cys-132(+7), Cys-135(+7), Tyr-139(+7)) and/or substrate binding and 2) conserved amino acid residues in VKORC1L1 (i.e., Lys-129(+7)), but not in VKORC1 (i.e., Tyr-129) that may cause a different susceptibility to VKAs between enzymes. Our analysis was centered on the area surrounding the presumed VKORC1 catalytic site [25,26], in which mutations leading to the most severe resistances to VKA have been detected [3,17,27]. This area was also proposed as putative-warfarin binding site [7,28]. These highlighted residues were subjected to systematic site-directed mutagenesis and we assessed the functional consequences of these mutations.
The Cys-132(+7) and Cys-135(+7) are totally conserved between VKORC1L1 sequences. These cysteines were also previously shown to be evolutionarily conserved in all known VKORC1 sequences [29]. In VKORC1L1, both cysteines are also in a “CXXC” motif (Figure 2). This motif was shown to be implicated in redox reactions in VKORC1 [30,31] but also in other redox proteins [32]. Recently, from two crystal structures of bacterial homologs captured in different reaction states, the second cysteines of the motif was proposed to link covalently to the C1 of the quinone ring of vit K1>0 [33]. In agreement with these results, mutations of cysteines 132 and 135 in alanine in hVKORC1 lead to an inactivation of the enzyme. In hVKORC1L1, the replacement of Cys-132(+7) and Cys-135(+7) of the “CXXC” motif by Ala, a nonconservative amino acid residue, totally abolished the VKOR activity. These results suggest the involvement of Cys-132(+7) and Cys-135(+7) in the transfer of the redox power from VKORC1L1 to vitamin K epoxide, as already proposed for VKORC1 [30].
Besides Cys-132 and Cys-135, two other cysteines Cys-43 and Cy-51 are strictly conserved in VKORC1 proteins of vertebrates, Drosophila, plants, bacteria and archeae. In VKORC1L1 proteins, both cysteines are also strictly conserved. In bacterial VKOR homologues, both cysteines are present in the predicted luminal loop separating α-helices 1 and 2. These loop cysteines were proposed to reduce the CXXC motif [31,33]. For VKORC1 proteins, there is still a debate between Cys-43 and Cys-51 being involved in the internal electron transfer. Indeed substitutions of Cys-43 or/and Cys-51by non-conservative residues lead to contradictory results [30,34-36]. To address the functional contribution of both residues in VKORC1L1, Cys-43(+7) or Cys-51(+7) were replaced by Ala, a non-conservative residue. These mutations totally abolished activity of recombinant VKORC1L1 enzyme, suggesting a crucial role of these conserved residues in the internal electron transfer from the redox protein partner to the internal catalytic site, as suggested recently by Tie et al. [37].
A theoretical reaction mechanism for converting vitamin K>0 to K by VKOR enzymes was proposed by Silverman based on a chemical model study [38]. This mechanism was revisited later by a quantum chemical study [39]. The theoretical model implied the CXXC catalytic site and one nearby acidic amino acid residue involved in the protonation of the epoxide oxygen. Nevertheless, this proposed reaction mechanism was never experimentally confirmed even for VKORC1 enzymes. In hVKORC1L1, only two acidic amino acid residues Glu-130(+7) and Tyr-139(+7) are located close to the CXXC catalytic site. Both residues are totally conserved between VKORC1L1 proteins. To explore their respective role in the reaction mechanism of VKORC1L1, non-conservative and conservative mutations of these residues were performed.
The non-conservative substitution of Tyr-139(+7) by Ala in hVKORC1L1 totally abolished VKORC1L1 activity, while the conservative substitution of Tyr-139(+7) by Phe partially restored VKORC1L1 activity with a Km similar to wild type VKORC1L1.These results highlight the importance of an aromatic residue at this position. The Tyr-139(+7) may be involved in the substrate binding via stacking interaction. Nevertheless, the replacement of this residue by Phe at position 139(+7) dramatically impaired the Vmax and thus the catalytic efficiency. The Tyr-139(+7) of VKORC1L1 might be involved in the protonation of the reaction intermediate produced by catalysis, as was proposed for VKORC1 [40]. Finally, even if VKORC1L1 is naturally 30-fold more resistant to VKAs then VKORC1, the replacement of Tyr-139(+7) by Phe leads also, as described for VKORC1 [27,41], to an increase of Ki of VKORC1L1 towards VKAs. This result suggests a similarity of VKA-binding sites between VKORC1 and VKORC1L1 and the involvement of Tyr-139(+7) in the binding of VKA by VKORC1L1 as proposed for VKORC1 [28].
The non-conservative substitutions of Glu-130(+7) by Ala or His in hVKORC1L1 did not modify the VKOR activity. These results are thus not in favor of the involvement of Glu-130(+7) in the reaction mechanism of hVKORC1L1 and more precisely in the protonation of the epoxide oxygen. Nevertheless, while the substitution of Glu- 130(+7) by Ala or His does not modify VKOR activity, the replacement of the same Glu-130(+7) by Asp drastically decreases VKOR activity with an activity lower than 20% compared with that of the wild type enzyme. Therefore the catalytic efficiency of this mutant is extremely decreased while the enzyme-substrate interaction is unchanged (Km similar to that of the wild type enzyme). This loss of activity is very surprising because in VKORC1 proteins an Asp is present at this corresponding position. The weak activity of VKORC1L1-E130(+7)D is potentially due to a modification of the steric hindrance of the CXXC catalytic site, leading to a change in the positioning of the substrate in the catalytic site. In view of these surprising results, we then evaluated the susceptibility of the VKORC1L1-E130(+7)A, -E130(+7)H, -E130(+7)D to VKAs. Indeed, the domain surrounding the CXXC motif from Phe-131 to Thr- 137 was recently proposed as putative warfarin-binding site for VKORC1 [27]. While VKORC1L1 is naturally very resistant to VKA (30-fold than VKORC1 towards warfarin), the simple substitution of Glu-130(+7) by Ala, His or Asp (i.e., the corresponding amino acid found in VKORC1) drastically modified susceptibility to VKAs. Their susceptibility to VKAs becomes similar to that of VKORC1. These results suggest that the natural resistance to VKA of VKORC1L1 is partially due to the steric hindrance of VKA interaction site by the lateral chain of Glu-130(+7). Nevertheless, the replacement of Asp-130 by Glu in VKORC1 does not modify the susceptibility of VKORC1 to VKA (data not shown), suggesting structural properties different between VKORC1 and VKORC1L1.
Conclusion
In conclusion, this structure-function study allowed us, 1) to demonstrate the importance of Cys-132(+7) and Cys-135(+7), but also of Cys-43(+7) and Cys-51(+7) in the transfer of the redox power from VKORC1L1 to vitamin K epoxide, 2) to compare the susceptibility to all VKAs of VKORC1 and VKORC1L1, 3) to demonstrate the implication of Tyr-139(+7) in the interaction of VKORC1L1 with warfarin, 4) to associate the natural resistance of VKORC1L1 to VKA with the steric hindrance of VKA interaction site by the lateral chain of Glu-130(+7). Interestingly, a single mutation is able to modify drastically the susceptibility of VKORC1L1 to VKAs.
Acknowledgment
This work was supported by grants from Agence Nationale pour la Recherche (RODENT 2009-CESA-008-03) and by DGER.
References
- Bell RG, Matschiner JT (1970) Vitamin K activity of phylloquinone oxide. Arch Biochem Biophys 141: 473-476.
- Li T, Chang CY, Jin DY, Lin PJ, Khvorova A, et al. (2004) Identification of the gene for vitamin K epoxide reductase. Nature 427: 541-544.
- Rost S, Fregin A, Ivaskevicius V (2004) Mutations in VKORC1 cause warfarin resistance and multiple coagulation factor deficiency type 2. Nature 427: 537-541.
- Silverman RB (1980) A model for a molecular mechanism of anticoagulant activity of 3-substituted 4-hydroxycoumarins. J Am Chem Soc 102: 5421-5423.
- Hodroge A, Matagrin B, Moreau C, Fourel I, Hammed A, et al. (2012) VKORC1 mutations detected in patients resistant to vitamin K antagonists are not all associated with a resistant VKOR activity. J Thromb Haemost 10: 2535-2543.
- Tie JK, Jin DY, Tie K, Stafford DW (2013) Evaluation of warfarin resistance using transcription activator-like effector nucleases-mediated vitamin K epoxide reductase knockout HEK293 cells. J Thromb Haemost11: 1556-1564.
- Czogalla KJ, Biswas A, Wendeln AC, Westhofen P, Müller CR, et al. (2013) Human VKORC1 mutations cause variable degrees of 4-hydroxycoumarin resistance and affect putative warfarin binding interfaces. Blood 122: 2743-2750.
- Westhofen P, Watzka M, Marinova M, Hass M, Kirfel G, et al. (2011) Human vitamin K 2,3-epoxide reductase complex subunit 1-like 1 (VKORC1L1) mediates vitamin K-dependent intracellular antioxidant function. J Biol Chem 286: 15085-15094.
- Oldenburg J, Watzka M, Bevans CG (2015) VKORC1 and VKORC1L1: why do vertebrates have two vitamin K2,3-epoxides reductases? Nutrients 7: 6250-6280.
- Hammed A, Matagrin B, Spohn G, Prouillac C, Benoit E, et al. (2013) VKORC1L1, an enzyme rescuing the VKOR activity in some extrahepatic tissues during anticoagulation therapy. J Biol Chem 288: 28733-28742.
- Price PA, Urist MR, Otawara Y (1983) Matrix Gla protein, a new gamma-carboxyglutamic acid-containing protein which is associated with the organic matrix of bone. Biochem Biophys Res Commun 117: 765-771.
- Price PA, Poser JW, Raman N (1976) Primary structure of the gamma-carboxyglutamic acid-containing protein from bovine bone. Proc Natl Acad Sci USA 73: 3374-3375.
- Nakano T, Kawamoto K, Kishino J, Nomura K, Higashino K, et al. (1997) Requirement of gamma-carboxyglutamic acid residues for the biological activity of Gas6: contribution of endogenous Gas6 to the proliferation of vascular smooth muscle cells. Biochem J 323: 387-392.
- Schurgers LJ, Teunissen KJ, Knapen MH, Kwaijtaal M, Van Diest R, et al. (2005) Novel conformation-specific antibodies against gamma-carboxyglutamic acid (Gla) protein: undercarboxylated matrix Gla protein as marker for vascular calcification. Arterioscler Thromb Vasc Biol 25: 1629-1633.
- Lee NK, Sowa H, Hinoi E, Ferron M, Ahn JD, et al. (2007) Endocrine regulation of energy metabolism by the skeleton. Cell 130: 456-469.
- Ferron M, Hinoi E, Karsenty G, Ducy P (2008) Osteocalcin differentially regulates beta cell and adipocyte gene expression and affects the development of metabolic diseases in wild-type mice. Proc Natl Acad Sci USA 105: 5266-5270.
- Hodroge A, Longin-Sauvageon C, Fourel I, Benoit E, Lattard V (2011) Biochemical characterization of spontaneous mutants of rat VKORC1 involved in the resistance to antivitamin K anticoagulants. Arch Biochem Biophys 515: 14-20.
- Ellis DJ, Usman MH, Milner PG, Canafax DM, Ezekowitz MD (2009) The first evaluation of a novel vitamin K antagonist, tecarfarin (ATI-5923) in patients with atrial fibrillation. Circulation 120: 1029-1035.
- Lasseur R, Longin-Sauvageon C, Videmann B, Billeret M, Berny P, et al. (2005) Warfarin resistance in a French strain of rats. J Biochem Mol Toxicol 19: 379-385.
- Grandemange A, Kohn MH, Lasseur R, Longin-Sauvageon C, Berny P, et al. (2009) Consequences of the Y139F Vkorc1 mutation on resistance to AVKs: in vivo investigation in a 7th generation of congenic Y139F strain of rats. Pharmacogenet Genomics 19: 742-750.
- Fregin A, Czogalla KJ, Gansler J, Rost S, Taverna M, et al. (2013) A new cell culture-based assay quantifies vitamin K2,3-epoxide reductase complex subunit 1 function and reveals warfarin resistance phenotypes not shown by the dithiotreitol-driven VKOR assay. J Thromb Haemost 11: 872-880.
- Le Heuzey JY, Ammentorp B, Darius H, De Caterina R, Schilling RJ, et al. (2014) Differences among western European countries in anticoagulation management of atrial fibrillation in Europe. Thromb Haemost 111: 833-841.
- Bavisotto LM, Ellis DJ, Milner PG, Combs DL, Irwin I, et al. (2011) Tecarfarin, a novel vitamin K reductase antagonist, is not affected by CYP2C9 and CYP3A4 inhibition following concomitant administration of fluconazole in healthy participants. J Clin Pharmacol 51: 561-574.
- Fourel I, Hugnet C, Goy-Thollot I, Berny P (2010) Validation of a new liquid chromatography-tandem mass spectrometry ion-trap technique for the simultaneous determination of thirteen anticoagulant rodenticides, drugs, or natural products. J Anal Toxicol 34: 95-102.
- Wajih N, Sane DC, Hutson SM, Wallin R (2005) Engineering of a recombinant vitamin K-dependent gamma-carboxylation system with enhanced gamma-carboxyglutamic acid forming capacity: evidence for a functional CXXC redox center in the system. J Biol Chem 280: 10540-10547.
- Tie JK, Nicchitta C, Von Heijne G, Stafford DW (2005) Membrane topology mapping of vitamin K epoxide reductase by in vitro translation/cotranslocation. J Biol Chem 280: 16410–16416.
- Watzka M, Geisen C, Bevans CG, Sittinger K, Spohn G, et al. (2011) Thirteen novel VKORC1 mutations associated with oral anticoagulant resistance: insights into improved patient diagnosis and treatment. J Thromb Haemost 9: 109-118.
- Oldenburg J, Bevans CG, Muller CR, Watzka M (2006) Vitamin K epoxide reductase complex subunit 1 (VKORC1): the key protein of the vitamin K cycle. Antioxid Redox Signal 8: 347-353.
- Goodstadt L, Ponting CP (2004) Vitamin K epoxide reductase: homology, active site and catalytic mechanism. Trends Biochem Sci 29: 289-292.
- Jin DY, Tie JK, Stafford DW (2007) A topological study of the human gamma-glutamyl carboxylase. Biochemistry 46: 7279-7283.
- Li W, Schulman S, Dutton RJ, Boyd D, Beckwith J, et al. (2010) Structure of a bacterial homologue of vitamin K epoxide reductase. Nature 463: 507-512.
- Holmgren A (1985) Thioredoxin. Annu Rev Biochem 54: 237-271.
- Liu S, Cheng W, Fowle Grider R, Shen G, Li W (2014) Structures of an intramembrane vitamin K epoxide reductase homolog reveal control mechanisms for electron transfer. Nat Commun 5: 3110.
- Rost S, Fregin A, Hünerberg M, Bevans CG, Müller CR, et al. (2005) Site-directed mutagenesis of coumarin-type anticoagulant-sensitive VKORC1: evidence that highly conserved amino acids define structural requirements for enzymatic activity and inhibition by warfarin. Thromb Haemost 94: 780-786.
- Rishavy MA, Usubalieva A, Hallgren KW, Berkner KL (2011) Novel insight into the mechanism of the vitamin K oxidoreductase (VKOR): electron relay through Cys43 and Cys51 reduces to allow vitamin K reduction and facilitation of vitamin K-dependent protein carboxylation. J Biol Chem 286: 7267-7278.
- Tie JK, Jin DY, Stafford DW (2012) Human vitamin K epoxide reductase and its bacterial homologue have different membrane topologies and reaction mechanisms. J Biol Chem 287: 33945-33955.
- Tie JK, Jin DY, Stafford DW (2014) Conserved loop cysteines of vitamin K epoxide reductase complex subunit 1-like 1 (VKORC1L1) are involved in its active site regeneration. J Biol Chem 289: 9396-9407.
- Silverman RB (1981) Chemical model studies for the mechanism of vitamin K epoxide reductase. J Am Chem Soc 103: 5939-5941.
- Davis CH, Deerfield D, Wymore T, Stafford DW, Pedersen LG (2007) A quantum chemical study of the mechanism of action of vitamin K epoxide reductase. J Mol Graph Model 26: 401-408.
- Matagrin B, Hodroge A, Montagut-Romans A, Andru J, Fourel I, et al. (2013) New insights into the catalytic mechanism of vitamin K epoxide reductase (VKORC1)-the catalytic properties of the major mutations of rVKORC1 explains the biological cost associated to mutations. FEBS Open Bio 3: 144-150.
- Pelz HJ, Rost S, Hünerberg M, Fregin A, Heiberg AC, et al. (2005) The genetic basis of resistance to anticoagulants in rodents. Genetics 170: 1839-1847.

