Keywords
Reactive oxygen species (ROS); Sepsis; Cardiac dysfunction; Endothelium; Microvascular dysfunction; Biomarker
Introduction
The development of sepsis is a critical challenge faced by physicians as the cumulative effect of sepsis-associated organ failure results in up to 70% mortality in the setting of severe hypotension [1]. In the United States, there are approximately 750,000 cases of severe sepsis diagnosed each year [2, 3]. Sepsis is defined as the presence of a systemic inflammatory response syndrome (SIRS) in response to an infection [4]. SIRS is defined as the presence of 2 or more of the following characteristics: 1) temperature > 38ºC or < 36 ºC, 2) heart rate > 90 beats/minute, 3) respiratory rate > 20 breaths/minute or PaCO2 < 32 torr (< 4.2 kPa), and 4) white blood cell count <4,000 or > 12,000 cells/mm3 or > 10% immature (band) forms [4]. Severe sepsis is the presence of sepsis associated with organ dysfunction, hypo-perfusion or hypotension (systolic blood pressure < 90 mmHG or reduction of > 40 mmHG from baseline in the absence of other causes for hypotension). [4]. Septic Shock is the presence of sepsis with hypotension despite adequate fluid resuscitation along with the presence of perfusion abnormalities [4]. Multiple organ dysfunctions describes the presence of altered organ function in an acutely ill patient such that homeostasis cannot be obtained without intervention [4].
Sepsis commonly induces cardiac dysfunction in critically ill patients, and approximately 50% of patients with sepsis show signs of cardiac dysfunction. These patients have a higher mortality rate than those patients with sepsis without cardiac dysfunction [5]. In fact, a patient’s ability to recover from cardiac dysfunction during sepsis is an important predictor of survival.
The underlying mechanisms which lead to the development of cardiac dysfunction in the setting of sepsis are not fully known. However, it has been demonstrated that factors involved include circulatory and micro vascular changes, contractile dysfunction, autonomic dysregulation, metabolic changes, increase in reactive oxygen species (ROS), mitochondrial dysfunction, cell death and inflammatory signaling [1, 6, 7].
Importantly, the circulatory and microvascular changes seen in sepsis are a natural part of the host response to infection. Although initially they are compensatory in nature, the circulatory and microvascular changes may become excessive resulting in cardiac dysfunction [8]. Cardiac dysfunction in the setting of sepsis is associated with abnormal oxygen delivery, macro and microcirculatory dysfunction, altered leukocyte-endothelium interactions, and increased formation of reactive oxygen species (ROS).
In this article, we aim to review the endothelial abnormalities associated with cardiac dysfunction in the setting of sepsis.
Cardiac Dysfunction in Sepsis
As described by Nasu and Sato cardiac dysfunction in sepsis is associated with three main characteristics: (i) left ventricular dilatation, (ii) depressed ejection fraction, and (iii) recovery after 7-19 days [9]. The left ventricular dilatation associated with sepsis was first described by Parker et al in 1984 [10]. This left ventricular dilatation is the result of left ventricular compliance and is associated with low filling pressure. The second characteristic of cardiac dysfunction associated with sepsis is depressed ejection fraction. Left ventricular dilatation leads to a decreased ability to contract leading to the depressed ejection fraction seen in septic patients [9-13]. Recent studies demonstrate the presence of right ventricular dilatation accompanied with decreased right ventricular function in sepsis as well. Severe sepsis is usually associated with both left and right ventricular dysfunction. In addition, left ventricular nondilatation, right ventricular dysfunction and diastolic dysfunction are associated with a worse prognosis [1, 9, 11, 14-16]. Finally, the third characteristic of cardiac dysfunction associated with sepsis is the heart’s ability to correct its dysfunction after 7-10 days [10-12, 17] (Figure 1).
Abnormal Cardiac Oxygen Delivery and Oxygen Consumption in Sepsis
Abnormal delivery-dependent oxygen consumption is a characteristic of cardiac dysfunction in sepsis [18]. This deficit may be caused: 1) by abnormal microcirculatory blood flow leading to cellular hypoxia, 2) from defects in the energy producing metabolic pathways in the cardiac cells, or 3) from a combination of both of these abnormalities. In patients with sepsis, oxygen consumption increases as cardiac output increases. In contrast, in healthy patients, oxygen consumption reaches a plateau and remains constant despite continuing increases in oxygen delivery. It is thought that in sepsis, delivery-dependent oxygen uptake signifies an underlying cellular deficiency in oxygen, making inadequate oxygen delivery an important factor in the pathogenesis of organ failure in septic patients [7, 19, 20]. Sepsis is known to cause increased oxygen consumption in cardiac tissue and to cause decreased coronary perfusion [21]. While fluid resuscitation can reverse the discrepancy between oxygen delivery and demand in early sepsis, this difference becomes difficult to overcome as patients deteriorate. Increased oxygen delivery to cardiac tissue is an ongoing struggle for the physician managing patients in cardiac dysfunction from sepsis.
Preserved Macrocirculatory Cardiac Blood Flow in Sepsis
Interestingly, coronary blood flow is increased in patients with septic shock. It is believed that myocardial tissue contains mechanisms of autoregulation that lead to a narrowed oxygen content difference (arterial minus coronary sinus) and diminished fractional extraction of arterial oxygen in the setting of sepsis. These findings suggest that global cardiac ischemia may not be the cause of cardiac dysfunction in septic shock [18, 22, 23]. This may also explain why patients with severe cardiac dysfunction in the setting of septic shock eventually do not respond to increased fluid resuscitation; rather they often require inotropic support.
Cardiac Microvascular Dysfunction in Sepsis
Microvascular dysfunction plays a central role in cardiac dysfunction. Vascular dysfunction affects all branches of the vascular system including the arterioles, capillaries and venules. Arteriolar dysfunction leads to abnormal tissue perfusion and is characterized by the loss of vasoreactivity [24-26]. Capillary dysfunction leads to the inability of oxygen delivery to the mitochondria of the cardiomyocyte and is characterized by decreased ability of cardiac tissue to extract oxygen [24, 25]. Venular dysfunction leads to aberrant inflammatory cell trafficking and protein exchange, and is characterized by enhanced neutrophil infiltration and protein leakage in the tissues [24, 25].
Research suggests that inflammation affects the coronary arteriolar response to both alpha (phenylephrine, clonidine) and beta adrenergic stimulation (isoproterenol) and to products released from platelets (ADP and serotonin) [27-29].
Chronic sepsis results in reduced alpha 2-adrenoreceptormediated relaxation of coronary arterioles [27]. In a rat model of chronic sepsis, subepicardial coronary arterioles were isolated and found to have reduced contractile responses to the protein kinase C activator (12-deoxyphorbol 13-isobutyrate 20-acetate) and the alpha 1-adrenoreceptor agonist (phenylephrine) [27]. In addition relaxation responses of the subepicardial coronary arterioles were reduced in response to the endothelium-dependent vasodilator (adenosine 5-diphosphate), the alpha 2-adrenorecetor agonist (clonidine), and the PKC inhibitor (staurosporine). However the relaxation to the endothelium-independent cyclic GMP-mediated vasodilator (sodium nitroprusside) was not affected. Relaxation to clonidine was inhibited by endothelial denudation or after blocking nitric oxide synthase. Together, these results suggested that sepsis significantly inhibited endothelium-dependent coronary vasorelaxation.

Figure 1: Pathway leading from sepsis to cardiac dysfunction as a result of endothelial dysfunction.
Systemic Inflammatory Response Syndrome (SIRS) in the presence of an infection constitutes sepsis. Initially, blood flow to the heart is preserved by autonomic regulation and tachycardia. At a later persistent inflammatory state of sepsis, there is an increase in endothelial ROS that leads to microvascular endothelial dysfunction. Altered endothelium-leukocyte interaction further exacerbates microvascular dysfunction and ROS production, which act as a feed-forward loop in sepsis. Together, these result in unequal oxygen delivery to and demand in the septic myocardial tissue. Increase in ROS (e.g. superoxide, peroxynitrite) generation in endocardium contributes to abnormal myocardial contractility and eventual cardiac dysfunction. Candidate biomarkers are indicated: soluble fms-like tyrosine kinase/sVEGFR1 (sFlt-1), plasminogen activator inhibitor-1 (PAI-1), sE-selectin, soluble intercellular adhesion molecule (sICAM-1), and soluble vascular cell adhesion molecule (sVCAM-1).
Beta 2-adrenoreceptors appear to affect coronoary microcirculation over beta 1-adrenoreceptors in a pig model of endotoxemia [29]. Preconstricted coronary arterioles dilated in response to either the Gs-protein activator (sodium fluoride), the adenylate cyclase activator (forskolin) or the nonselective beta-adrenoreceptor agonist (isoproterenol). After 3 hours of endotoxemia, the relaxation response to sodium fluoride and isoproterenol was significantly reduced; however, the response to forskolin was not changed. Addition of a beta 2-adrenoceptor blocker, (ICI-118, 551) reduced the relaxation of the control microvessels induced by isoproterenol while the beta 1-adrenoceptor blocker (atenolol) only slightly reduced the isoproterenol-induced relaxation [29]. Taken together, these data indicate that sepsis impairs beta 2-adrenoceptor-mediated relaxation in the porcine coronary microcirculation.
Sepsis also alters the reaction of coronary vessels to platelet products [28]. A pig model of endotoxemia was used to study the effect of sepsis on coronary and pulmonary responses to platelet products, e.g. serotonin and ADP [28]. Coronary arterioles from the sepsis pigs were contracted in the presence of serotonin which was reversed by indomethacin [28]. The relaxation response was more significant in the septic pigs than in the control pigs. In contrast, relaxation response to ADP was unchanged in endotoxemia [28]. Similar results were seen in the pulmonary arterioles [28]. Relaxation induced by sodium [28] nutriprusside (SNP) was unaffected by endotoxemia in both the coronary and pulmonary arterioles. These data suggest that coronary microvascular relaxation responses to serotonin are significantly reduced, even are converted to contractile responses in endotoxemia while the relaxation response to ADP and SNP is minimally affected by endotoxemia [28]. Together, the findings pointed towards a dysfunctional microvascular response to the platelet-specific product serotonin in porcine model of sepsis.
Interestingly, Thijs et al. used a canine model of endotoxin induced shock to investigate whether heterogeneous coronary blood flow is distributed evenly throughout cardiac tissue [23]. Using radioactive microspheres to study myocardial blood flow in small tissue sections, these authors evaluated hemodynamic variables and global myocardial metabolism. They reported that heterogeneous blood flow was distributed unevenly in areas of the heart while global blood flow was well-maintained. This flow-micro flow misdistribution may well be associated with focal areas of ischemia which could contribute to the overall cardiac dysfunction seen in the setting of sepsis [23]. Sepsis has also been found to lead to microvascular dysfunction by causing swelling of endothelial cells and increasing non-occlusive intravascular fibrin deposition in the heart [7, 20]. Therapies aimed at improving the microvascular dysfunction associated with sepsis may provide an effective means to reverse cardiac dysfunction.
Enhanced Leukocyte and Endothelium Interactions in Sepsis
The leukocyte-endothelium interaction is an important aspect leading to microvascular disease. The inflammation associated with sepsis leads to vascular leakage and myocardial edema resulting in reduced cardiac function and compliance [7]. In the presence of inflammatory stimuli, vascular endothelium directs leukocytes to sites of vascular injury. In sepsis, there is an increase in leukocyte rolling and adhesion which leads to the production of toxic mediators by the dysfunctional endothelium and the activated leukocytes [30, 31].
In the setting of sepsis endocardium is known to 1) have a pro-inflammatory phenotype, 2) contain elevated levels of proinflammatory transcription factor, such as NFkB, [24, 32], 3) induce the expression of adhesion molecules on endothelial cells [33], 4) promote adhesion (and thus slow rolling) of PMN cells to endothelial cells (EC) [32, 34], 5) promote trans-endothelial migration of polymorphonuclear (PMN) cells into the cardiac insterstitium [35], and 6) promote the adhesion of PMN cells to cardiomyoctyes. These endothloelium-adhered PMN cells induce an increase in oxidative stress in ECs and cardiomyocytes, which impair cardiac contractility [32, 34-40].
Kviety et al studied the mechanisms involved in cardiac myoctye activation in sepsis and the means by which the activated myoctytes promote PMN trans endothelial migration. They found that TNF-α and IL-1 help activate cardiac myoctytes and that various chemokines (including LIX, KC and platelet activating factor) cause the activated myocytes to promote PMC transendothelial migration [35].
Calpain has been found to be over activated in cardiac tissue in the setting of sepsis induced inflammation. Neviere et al investigated the effects of calpain inhibition on myocardial dysfunction and inflammation in a rat model of endotoxin-induced sepsis [6]. In animal studies, septic mice had reduced systolic function which was partially improved by the addition of calpain inhibitors [6, 41]. In one study, calpain inhibition reduced plasma levels of TNF alpha and nitrite / nitrate levels in the septic rats. In their animal model, leukocyte rolling on and adhesion to the venular endothelium was increased. This increased leukocyteendothelium interaction was reversed by calpain inhibitors. The attenuation of leukocyte-endothelium interactions observed in the septic rats treated with calpain inhibitor was associated with increased levels of the plasma anti-adhesion molecule, e.g. endocan (ESM-1). Finally the hearts from the endotoxin treated rats had reduced systolic function that was improved with the addition of a calpain inhibitor [6]. In another study, calpain activation in lipopolysaccharide (LPS)-induced cardiomycytes was found to activate caspase 3 and TNFα. Another study reported that calpain inhibition improved myocardial function in a mouse model of endotoxaemia [41]. Together, these findings suggested that calpain inhibition improved sepsis-induced cardiac dysfunction by attenuating endothelium-leukocyte interactions on the inflamed endothelium [6, 31, 41].
In addition, increased expression of vascular cell adhesion molecule (VCAM-1) and neutrophil accumulation are seen in myocardial tissue in a mice model of lipopolysaccharide (LPS)- induced sepsis and myocardial dysfunction. Blockade of VCAM-1 abrogates neutrophil accumulation in the myocardial tissue and preserves cardiac function. This suggests that VCAM-1 may serve as a therapeutic target for myocardial protection during sepsis [33, 42].
Therapies aimed at attenuating increased leukocyte-endothelium interaction seen in sepsis may provide a promising means of providing cardiac protection.
Reactive Oxygen Species in Sepsis
Reactive oxygen species (ROS) are molecules that have oxidizing ability. ROS may or may not have free electrons. Examples of ROS include superoxide (O2⋅-), hydroxyl anion (HO⋅), and nitric oxide (NO⋅), hydrogen peroxide (H2O2), hypochlorous acid (HOCl), and peroxynitrite (ONOO-). At physiological concentrations ROS are essential for normal signal transduction in endothelial cells. Overproduction of ROS or reduced availability of antioxidant enzymes have traditionally been considered to play an essential role in the pathogenesis of cardiovascular dysfunction [43, 44].
Sys et al investigated the role of endothelial paracrine regulation of cardiac function in the setting of sepsis [44]. Specifically, the authors investigated the role of cardiac endothelium-derived endothelin-1, prostaglandin and nitric oxide (NO) during endotoxin-induced cardiomyopathy in rabbits [39]. They found an increase in NO synthase (eNOS) and cycloocygenase-2 in the endocardium and coronary arteriolar endothelium in the septic myocardial tissue. Increase in NO may initially act to compensate for the increased ROS levels in septic endocardium and myocardium. However, increase in ROS (specifically superoxide) and NO may finally lead to increase in ONOO. Nitrotyrosine was similarly increased in the endothelial tissues in these septic hearts. Contractile function of the papillary muscles was depressed and the isometric twitches were prolonged. This twitch prolongation was reversed by the administration of drugs that blocked endothelin-1 and prostaglandin. Interestingly, in the septic group, myocardial inotropic responsiveness to endothelin-1 was increased. Therefore, it appears that coronary endothelial activation during sepsis leads to sensitization of myocardial tissue to endothelium-derived cytokine and inflammatory mediators [45-47].
A mechanism by which increased ROS released from the endothelium leads to cardiac dysfunction in sepsis has been suggested. It is thought that the nitric oxide that is released from activated endothelial cells in the setting of sepsis causes dysfunctional isometric contractions of cardiac trabeculae leading to cardiac dysfunction [7]. It has been shown that in septic conditions, lysozymes bind to endocardium which generates nitric oxide production. This nitric oxide then activates the myocardial guanosine 3’5’ monophosphate pathway which leads to cardiac depression [48, 49].
However, recent data support the notion that increased ROS levels may have a protective role in vascular endothelium and may lead to improved coronary endothelial function [50-52]. For example, in the setting of an inflammatory insult, vascular stress leads to the elevated levels of nitric oxide, which in turn has mitochondrial protective functions and leads to mitochondrial biogenesisis; this may serve as a cardiac repair mechanism [44].
Taken together, ROS play intriguing roles in sepsis-induced cardiac dysfunction, ranging from beneficial to deleterious effects. It is likely that the sub-cellular sources of ROS including spatial and temporal changes in the ROS levels in endothelium determine specific cardiovascular outcomes in sepsis (Figure 2).
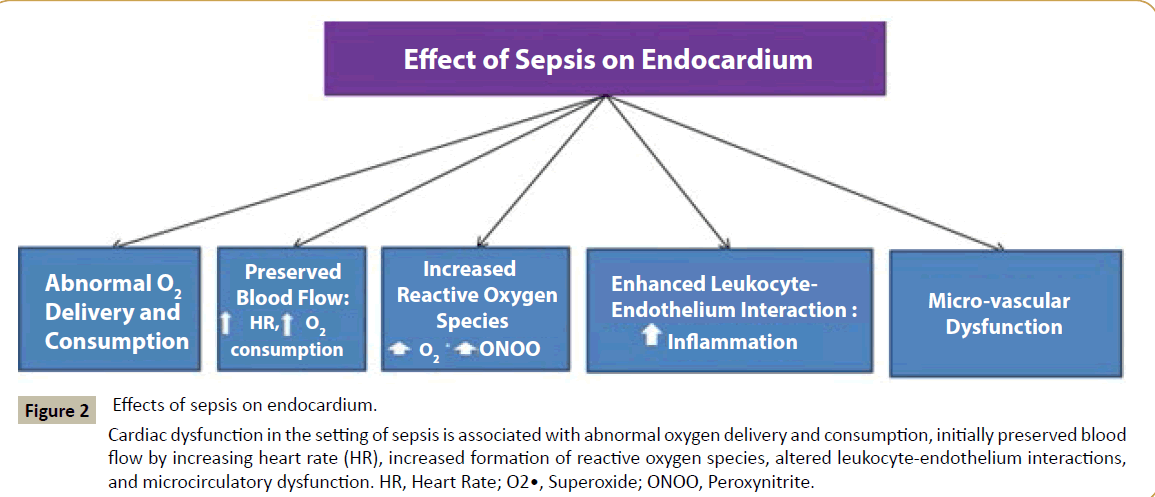
Figure 2:Effects of sepsis on endocardium.
Cardiac dysfunction in the setting of sepsis is associated with abnormal oxygen delivery and consumption, initially preserved blood flow by increasing heart rate (HR), increased formation of reactive oxygen species, altered leukocyte-endothelium interactions, and microcirculatory dysfunction. HR, Heart Rate; O2•, Superoxide; ONOO, Peroxynitrite.
Clinical Biomarkers of Cardiac Dysfunction
Early detection of cardiac dysfunction in sepsis has significant clinical importance. Specific biomarkers of cardiac dysfunction in the setting of sepsis will help detect early indication of myocardial dysfunction and help monitor critical cases. Plasma troponins are currently used to estimate the presence of damage to myocyte cell membrane integrity. The levels of troponin in the plasma can be affected significantly by acute myocardial ischemic injury, increased myocardial stress or demand and the inability to clear troponin in patients with low renal function [9]. B-type natriuretic peptide (BNP) plasma levels are used to estimate the presence of myocardial wall stress [53]. However, BNP plasma values increase in the setting of other critical illnesses rather than just sepsisinduced cardiac dysfunction [54, 55]. Neither Troponin nor BNP are specific biomarkers of cardiac dysfunction in the setting of sepsis.
In patients who present to the emergency department with a clinical suspicion of infection, biomarkers of endothelial activation have been found to be associated with sepsis severity, the amount of organ dysfunction and mortality. These biomarkers include soluble fms-like tyrosine kinase (sFlt-1), plasminogen activator inhibitors-1 (PAI-1), sE-selectin, soluble intercellular adhesion molecule (sICAM-1) and soluble vascular cell adhesion molecule (sVCAM-1) [2, 8]. In addition, the presence of endothelin-1 has been found to be associated with both left ventricular and right ventricular dysfunction in the setting of sepsis [1]. Taken together, these findings suggest that improved understanding of endothelial dysfunction associated with sepsis may lead to novel endothelium directed therapies.
Clinical Relevance, Advantages and Limitations of the Current Review
In this review article, we briefly discussed the clinical challenges and our current understanding of the endothelial dysfunction associated with cardiac dysfunction in sepsis. We focused mainly on the pathological changes that occur in vascular endothelium, with an emphasis on endocardium, and how endothelial ROS, impaired endothelium-leukocyte interaction, and microcirculatory changes lead to cardiac dysfunction in sepsis.
A limitation of our review is that it does not look at the inflammatory markers and other parameters that may be contributing to cardiac dysfunction during sepsis. However, poor blood flow and impaired oxygen delivery and utilization are important components of organ dysfunction in sepsis. This current review has focused mainly on the changes that occurred in the endothelium during sepsis and thus provides insight into potential therapies that may help to reverse the endothelial dysfunction and potentially treat sepsis-induced cardiac dysfunction.
Limitations of the Previous Studies
The above literature shows that in the setting of sepsis, there is dysfunction of the coronary endothelial cells that leads to cardiac dysfunction. The research suggest that that sepsis leads to increase in ROS, microvascular dysfunction, and enhanced leukocyte-endothelial interaction- all causing dysfunctional endothelium leading to impaired oxygen delivery and impaired myocardial oxygen consumption. This eventually leads to sepsisinduced cardiac dysfunction. This pathway has been well and thoroughly studied.
However, there is a conspicuous lack of research showing how endothelial dysfunction associated with sepsis actually causes the three main characteristics of cardiac dysfunction as described by: (i) left ventricular dilatation, (ii) depressed ejection fraction, and (iii) recovery after 7-19 days. One explanation that has been offered is ROS. However, the precise mechanisms are not yet elucidated about how ROS bring about these diverse changes leading to cardiac dysfunction. One theory is that the coronary endothelial activation (that occurs via increases in ROS) during sepsis leads to sensitization of myocardial tissue to endotheliumderived cytokine and inflammatory mediators [44, 45]. Further it has been shown that localized increase in nitric oxide (NO) that is released from activated endothelial cells in the setting of sepsis causes dysfunctional isometric contractions of cardiac trabecular leading to cardiac dysfunction. A differential distribution gradient of NO in the cardiac trabeculae has been suggested to be the reason behind the dysfunctional isometric contractions [7]. Future studies will need to evaluate these mechanisms more thoroughly.
Directions for Further Studies
Approximately 50% of patients with sepsis show signs of cardiac dysfunction. These patients have a higher mortality rate than those patients without cardiac dysfunction [5]. In fact, a patient’s ability to recover from cardiac dysfunction during sepsis is an important predictor of survival and this recovery usually occurs within 7-19 days. Research to date has identified many different cellular pathways in endocardium which are affected in sepsis. Future studies are needed to unlock the mechanisms that lead some patients to develop cardiac dysfunction in sepsis while others do not. In addition, it is critical to understand the mechanisms by which some patients recover from cardiac dysfunction around the critical period of 7-10 days, which will certainly help develop therapeutic modalities aimed at treating cardiac dysfunction in sepsis.
As mentioned earlier, there is a clear lack of effective biomarkers that could predict cardiac outcomes in sepsis. The current biomarkers for sepsis-induced cardiac dysfunction include endothilin-1, sFlt-1, PAI-1, sICAM-1 and sVCAM-1, which are not very specific [1, 2, 8]. Studies are underway to identify biomarkers associated with sepsis which will help predict or determine severity of organ dysfunction and mortality. Effective biomarkers also needed to help understand patients’ response to treatment aimed at cardiac dysfunction in sepsis. This review suggests a novel research direction towards endothelium-directed therapies for sepsis-induced cardiac dysfunction.
Conclusion
Cardiac dysfunction is a common and serious consequence of septic shock. Severe sepsis can lead to both right and left ventricular compromise which is associated with an increased mortality. In the setting of sepsis, there is an increase in oxygen consumption in cardiac tissue. Whereas global blood flow to the heart is preserved, it is the microvascular changes that occur in the setting of sepsis contributing to decreased cardiac function. Therefore, reversing the microvascular dysfunction or attenuating the enhanced leukocyte-endothelium interaction seen in sepsis provides promising mechanisms of treating and / or preventing cardiac dysfunction.
Sources of Funding
This work was supported by the National Institute of General Medical Sciences (NIGMS) / National Institute of Health (NIH) grant 1P20GM103652 (Project# 3) (to MRA) and American Heart Association (AHA) Grant-in-Aid 14GRNT20460291 (to MRA); National Heart, Lung, and Blood Institute (NHLBI) grant HL46716 (to FWS). BAP was supported by an NIH/NIGMS Training Grant 2T32 GM065085.
References
- Furian T, Aguiar C, Prado K, Ribeiro RV, Becker L, et al. (2012) Ventricular dysfunction and dilation in severe sepsis and septic shock: Relation to endothelial function and mortality. J Crit Care 27:e9-e15.
- Shapiro NI, Schuetz P, Yano K, Sorasaki M, Parikh SM, et al. (2010) The association of endothelial cell signaling, severity of illness, and organ dysfunction in sepsis. Crit Care 14:R182.
- Hochstadt A, Meroz Y, Landesberg G (2011) Myocardial dysfunction in severe sepsis and septic shock: More questions than answers? J CardiothoracVascAnesth 25:526-535.
- Committee M of the AC of CP of CCMCC (1992) American College of Chest Physicians/Society of Critical Care Medicine Consensus Conference: Definitions for Sepsis and Organ Failure and Guidelines for the Use of Innovative Therapies in Sepsis. Crit Care Med Care Med 20:864-874.
- Zaky A, Deem S, Bendjelid K, Treggiari MM (2014) Characterization of cardiac dysfunction in sepsis: an ongoing challenge. Shock 41:12-24.
- Tissier S, Lancel S, Marechal X, Mordon S, Depontieu F, et al. (2004) Calpain inhibitors improve myocardial dysfunction and inflammation induced by endotoxin in rats. Shock 21:352-357.
- Rudiger A, Singer M (2007) Mechanisms of sepsis-induced cardiac dysfunction. Crit Care Med 35:1599-1608.
- Skibsted S, Jones AE, Puskarich MA, Arnold R, Sherwin R, et al. (2013) Biomarkers of endothelial cell activation in early sepsis. Shock 39:427-432.
- Sato R, Nasu M (2015) A review of sepsis-induced cardiomyopathy. J Intensive Care 3: 1-7.
- Parker MM, Shelhamer JH, L BS (1984) Profund but reversible myocardial depression in patients with septic shock. Ann Intern Med 100: 483-490.
- Krishnagopalan S, Kumar A, Parrillo JE, Kumar A (2002) Myocardial dysfunction in the patient with sepsis. CurrOpinCrit Care 8:376-388.
- Jardin F, Fourme T, Page B, Loubières Y, Vieillard-Baron A, et al. (1999) Persistent preload defect in severe sepsis despite fluid loading: A longitudinal echocardiographic study in patients with septic shock. Chest 116:1354-1359.
- Moore TD, Frenneaux MP, Sas R, Atherton JJ, Morris-Thurgood JA, et al. (2001) Ventricular interaction and external constraint account for decreased stroke work during volume loading in CHF. Am J Physiol Heart CircPhysiol 281:H2385-H2391.
- Kimchi A, Ellrodt AG, Berman DS, Riedinger MS, Swan HJ, et al. (1984) Right ventricular performance in septic shock: A combined radionuclide and hemodynamic study. J Am CollCardiol 4:945-951.
- Takada K, Matsumoto S, Hiramatsu T, Kojima E, Shizu M, et al. (2009) Spontaneous pneumomediastinum: an algorithm for diagnosis and management.TherAdvRespir Dis 3: 301-307.
- Gustavo Rolando, EDV, Espinoza, Avid E, Welsh S, et al. (2015) Prognostic value of ventricular diastolic dysfunction in patients with severe sepsis and septic shock. Rev Bras TerIntensiva 27:333-339.
- Natanson C, Eichenholz PW, Danner RL, PQ Eichacker, W Hoffman, et al. (1989) Endotoxin and Tumor Necrosis Factor Challenges in Dogs Simulate the Cardiovascular Endotoxin , an LPS found in the outer membrane of Gram-negative bacteria, has been considered by many to be the principal toxin involved in the pathogenesis of Gram-negative. J Exp Med i:823-832.
- Robert EC, Gary LS, Margaret MP, Charles N, Joseph EP (2013) The Coronary circulation in human septic shock. Circulation 73:637-644.
- Hotchkiss RS, Karl IE (1992) Reevaluation of the role of cellular hypoxia and bioenergetic failure in sepsis. JAMA 267:1503-1510.
- Solomon MA, Correa R, Alexander HR, Koev LA, Cobb JP, et al. (1994) Myocardial energy metabolism and morphology in a canine model of sepsis. Am J Physiol 266:H757-H768.
- Chagnon F, Bentourkia M, Lecomte R, Lessard M, Lesur O, et al. (2006) Endotoxin-induced heart dysfunction in rats: assessment of myocardial perfusion and permeability and the role of fluid resuscitation. Crit Care Med 34: 127-133.
- Dhainaut JF, Huyghebaert MF, Monsallier JF, Lefevre G, Dall'Ava-Santucci J, et al. (1987) Coronary hemodynamics and myocardial metabolism of lactate, free fatty acids, glucose, and ketones in patients with septic shock. Circulation 75:533-541.
- Groeneveld AB, van Lambalgen AA, van den Bos GC, Bronsveld W, Nauta JJ (1991) Maldistribution of heterogeneous coronary blood flow during canine endotoxin shock. Cardiovasc Res 25:80-88.
- Lush CW, Kvietys PR (2000) Microvascular dysfunction in sepsis. Microcirculation 7: 83-101.
- Olivo G, Consales G, Michelagnoli G (2006) Sepsis associated cardiomyopathy. CurrAnaesthCrit Care 17:349-358.
- Cameron EM, Wang SY, Fink MP SF (1998) Mesenteric and skeletal muscle microvascular responsiveness in subacute sepsis. Shock 9:184-192.
- Wang SY, Cameron EM, Fink MP SF (1997) Chronic septicemia alters alpha-adrenergic mechanisms in the coronary circulation. J Surg Res 69:61-66.
- Sellke FW, Wang SY, VanderMeer TJ FM (1994) Escherichia coli endotoxemia alters coronary and pulmonary arteriolar responses to platelet products. Shock 1:279-285.
- Wang SY, VanderMeer TJ, Fink MP SF (1994) Uncoupling of coronary microvascular beta 2-adrenoceptors by Escherichia coli endotoxemia. Surgery 116:307-312.
- Stalker TJ, Skvarka CB, Scalia R (2003) A novel role for calpains in the endothelial dysfunction of hyperglycemia. FASEB J 17:1511-1513.
- Potz BA, Sabe AA, Abid MR, Sellke FW (2015) Calpains and Coronary Vascular Disease. Circ J. 80: 4-10.
- Neviere RR, Cepinskas G, Madorin WS, Hoque N, Karmazyn M, et al. (1999) LPS pretreatment ameliorates peritonitis-induced myocardial inflammation and dysfunction: role of myocytes. Am J Physiol 277:H885-H892.
- Raeburn CD, Calkins CM, Zimmerman MA (2001) Vascular cell adhesion molecule-1 expression is obligatory for endotoxin-induced myocardial neutrophil accumulation and contractile dysfunction. Surgery 130:319-325.
- Madorin WS, Cepinskas G, Kvietys PR (2001) Peritonitis induces rat cardiac myocytes to promote polymorphonuclear leukocyte emigration and activate endothelial cells: effect of lipopolysaccharide pretreatment. Crit Care Med 29:1774-1779.
- Madorin WS, Rui T, Sugimoto N,Handa O, Cepinskas G, et al. (2004) Cardiac Myocytes Activated by Septic Plasma Promote Neutrophil Transendothelial Migration: Role of Platelet-Activating Factor and the Chemokines LIX and KC. Circ Res 94:944-951.
- Paul HR, Christopher AW, Wayne RG PK (1997) Emigrated Rat Neutrophils Adhere to Cardiac Myocytes via a4 Integrin. Circ Res 81:196-201.
- Poon BY, Ward CA, Giles WR, Kubes P (1999) Emigrated neutrophils regulate ventricular contractility via alpha4 integrin. Circ Res 84:1245-1251.
- Youker K, Smith CW, Anderson DC, Miller D, Michael LH, et al. (1992) Neutrophil adherence to isolated adult cardiac myocytes. J Clin Invest 89:602-609.
- Smith CW, Entman ML, Lane CL, Beaudet AL, Ty TI, et al. (1991) Adherence of neutrophils to canine cardiac myocytes in vitro is dependent on intercellular adhesion molecule-1. J Clin Invest 88:1216-1223.
- Youker K, Smith CW, Anderson DC, Miller D, Michael LH, et al. (1992) Neutrophil adherence to isolated adult cardiac myocytes. J Clin Invest 89:602–609.
- Kacimi R, Karliner JS, Koudssi F, Long CS (1998) Expression and Regulation of Adhesion Molecules in Cardiac Cells by Cytokines. Circ Res 82:576-586.
- Li X, Li Y, Shan L, Shen E, Chen R, et al. (2009) Over-expression of calpastatin inhibits calpain activation and attenuates myocardial dysfunction during endotoxaemia. Cardiovasc Res 83:72-79.
- Raeburn CD, Calkins CM, Zimmerman MA,Song Y, Ao L, et al. (2002) ICAM-1 and VCAM-1 mediate endotoxemic myocardial dysfunction independent of neutrophil accumulation. Am J PhysiolRegulIntegr Comp Physiol 283:R477-86.
- Lee M, Choy WC, Abid MR (2011) Direct sensing of endothelial oxidants by vascular endothelial growth factor receptor-2 and c-Src. PLoS One 6: 1-7.
- Bartz RR, Suliman HB, Piantadosi CA, Bartz RR (2015) Redox mechanisms of cardiomyocyte mitochondrial protection. Front Physiol 6:1-8.
- Mebazaa A, De Keulenaer GW, Paqueron X, Andries LJ, Ratajczak P, et al. (2001) Activation of cardiac endothelium as a compensatory component in endotoxin-induced cardiomyopathy: role of endothelin, prostaglandins, and nitric oxide. Circulation 104: 3137-3144.
- Jacobs H, Mink SN, Duke K, Bose D, Cheng ZQ, et al. (2005) Characterization of membrane N-glycan binding sites of lysozyme for cardiac depression in sepsis. Intensive Care Med 31:129-137.
- Mink SN, Bose R, Roberts DE, Jacobs H, Duke K, et al. (2005) Lysozyme binding to endocardial endothelium mediates myocardial depression by the nitric oxide guanosine 3',5' monophosphate pathway in sepsis. J Mol Cell Cardiol 39:615-625.
- Abid MR, Sellke FW (2015) Antioxidant Therapy: Is it your Gateway to Improved Cardiovascular Health? Pharm Anal acta 6:1-10.
- Shafique E, Choy WC, Liu Y, Feng J, Cordeiro B, et al. (2013) Oxidative stress improves coronary endothelial function through activation of the pro - survival kinase AMPK. Aging (Albany NY) 5:515-530.
- Abid MR, Spokes KC, Shih S-C, Aird WC (2007) NADPH oxidase activity selectively modulates vascular endothelial growth factor signaling pathways. J BiolChem 282: 35373-35385.
- Feng J, Damrauer SM, Lee M, Sellke FW, Ferran C, et al. (2010) Endothelium-Dependent Coronary Vasodilatation Requires NADPH Oxidase – Derived Reactive Oxygen Species. ArterThrombVascBiol 30:1703-1710.
- Abid MR, Tsai JC, Spokes KC, Deshpande SS, Irani K, et al. (2001) Vascular endothelial growth factor induces manganese- superoxide dismutase expression in endothelial cells by a Rac1-regulated NADPH oxidase-dependent mechanism. FASEB J 15:1-22.
- Charpentier J, Luyt CE, Fulla Y, Vinsonneau C, Cariou A, et al. (2004) Brain natriuretic peptide: A marker of myocardial dysfunction and prognosis during severe sepsis. Crit Care Med 32:660-665.
- Papanikolaou J, Makris D, Mpaka M, Palli E, Zygoulis P, et al. (2014) New insights into the mechanisms involved in B-type natriuretic peptide elevation and its prognostic value in septic patients. Crit Care 18:R94.