Keywords
PCSK9; LDL receptor; Protein mis-folding; Protein secretion; AAV8 transfection
Introduction
Proprotein Convertase Subtilisin-like Kexin type 9 (PCSK9) is an attractive target for the treatment of dyslipidemia [1,2]. Human genetic data for PCSK9 indicates a positive correlation between reduced low density lipoprotein-cholesterol (LDL-c) levels and decreased mortality from coronary heart disease (CHD) [3,4]. Multiple studies, including the Dallas Heart Study, demonstrate PCSK9 gain of function (GOF) mutations lead to increased plasma LDL-c levels and early onset of myocardial infarction (MI), whereas loss of function (LOF) mutations exhibit the opposite behavior (decreased plasma LDL-c levels and reduced incidence of CHD) [5,6]. Late-stage clinical trials with alirocumab and evolocumab, monoclonal antibodies that specifically bind PCSK9 and block PCSK9 function, provide direct clinical validation for the LDL-c lowering effect of PCSK9 inhibition [7-9], leading to their recent approval for heterozygous familial hypercholesterolemia (HeFH) and patients with clinical atherosclerotic cardiovascular disease. The mechanism by which PCSK9 regulates plasma LDL-c levels has been extensively explored [1]. Briefly, plasma LDL-c binds to the low-density lipoprotein receptors (LDLR) in the liver and is internalized for degradation. PCSK9, immediately following translation in hepatocytes, undergoes autocatalytic cleavage, through which a 10 kDa prodomain is cleaved from the 63 kDa mature protein, remaining non-covalently bound. Following secretion, cleaved PCSK9 binds to LDLR, directing the PCSK9: LDLR complex to lysosomes for degradation, thus controlling the level of LDL-c in plasma. Consistent with this mechanism, x-ray studies of wild type (WT) PCSK9 co-crystallized with the epidermal growth factor-like repeat (EGF-A) domain of LDLR reveal that several human GOF mutations lie in the catalytic domain of PCSK9 [10] which forms direct contacts with the EGF-A domain.
In addition, the C-terminal domain (CTD) of PCSK9 has also been suggested to play a role in mediating LDLR degradation [11- 18]. Zhang et al. [15] found that PCSK9 C-terminal domain is essential for the stimulation of LDLR degradation even though it is not required for binding of PCSK9 to the LDLR. Holla et al. [11] and Tveten et al. [14] further concluded that both the positive charge and size of the PCSK9 C-terminal domain are critical for maintaining the binding of PCSK9 to LDLR during receptor translocation and degradation. Saavedra [17] dissected the function of three tightly packed modules (namely, M1, M2 and M3) of the CTD and demonstrated that the M2 module was critical for extracellular PCSK9 induced LDLR degradation, while the other two modules might play an essential trafficking role for the PCSK9-LDLR complex [18]. Furthermore, several naturally occurring PCSK9 mutants in the C-terminal domain have been identified. Among these mutants, S462P [19] and A522T [20], both located in the C-terminal domain loop interfacing with the catalytic domain, are LOF mutations.
While the non-synonymous polymorphisms S462P and A522T PCSK9 present with a loss of function phenotype in man, here we utilize in vitro approaches and controlled mouse studies to understand the cell biology underlying the phenotype. in vitro, S462P and A522T PCSK9 mutations impact the ability of cleaved PCSK9 to be secreted, likely due to mis-folding, as suggested by an increased association with calreticulin. in vivo, plasma hPCSK9 levels were significantly lower for mutant PCSK9’s leading to decreases in plasma cholesterol consistent with increased LDLR. Notably, these differences were due to altered secretion of hPCSK9 given that Pcsk9 transcript levels are equivalent in mouse liver. Molecular modeling indicates that S462P and A522T PCSK9 mutations destabilize the C-terminal domain of PCSK9 to varying degrees with the severity correlating with the impact on folding and secretion. These observations provide further insight into a different mechanism of LDL-c regulation wherein PCSK9 function is reduced by decreasing secretion instead of affecting LDLR binding or impacting its auto-cleavage.
Materials and Methods
Materials
Antibodies: PCSK9 monoclonal antibodies E07 and B20 were produced internally. All other antibodies were purchased from external vendors: Goat polyclonal antibody against PCSK9 (Abcam, Cambridge, MA, USA), mouse monoclonal antibody against actin (Sigma, St. Louis, MO, USA), rabbit polyclonal antibodies against LDLR (Abcam; Fitzgerald Industries International, Acton, MA, USA), mouse monoclonal antibody against V5 (Thermo Fisher Scientific, Waltham, MA, USA), rabbit polyclonal antibody against V5 (EMD Millipore, Temecula, CA, USA), mouse monoclonal antibody against calreticulin (Abcam). Rabbit IgG (Thermo Fisher Scientific) and mouse IgG2 (BD Biosciences, San Jose, CA, USA) were used as isotype controls. These secondary antibodies conjugated with IRDye were used for western blot (Licor, Lincoln, NE, USA): IRDye 800CW-goat-anti-mouse antibody, IRDye 680LTgoat- anti-rabbit antibody, and IRDye 800CW-donkey-anti-goat antibody.
Cell culture
Human Embryonic Kidney (HEK) 293 and HepG2 cells from American Type Culture Collection (ATCC, Manassas, VA, USA) were cultured in Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS, v/v), 100 μg/ml streptomycin sulfate and 100 units/ml penicillin (all from Thermo Fisher Scientific).
Construction and establishment of PCSK9 mutant stable lines
All the PCSK9 mutants were constructed based on the WT human PCSK9 plasmid reported previously [21]. Each mutation was introduced by polymerase chain reaction (PCR) and generated by Genewiz, Inc. (South Plainfield, NJ, USA).
All PCSK9 mutant stable cell lines were established in HEK293 cells. D374Y and S127R were established previously [22]. The other four mutants (S462A, S462P, A522T, A522G) were set up as following: HEK293 cells were seeded at 4 × 106 cells in a T75 flask (Thermo Fisher Scientific) and cultured in DMEM containing 100 units of penicillin and 100 mg/ml streptomycin sulfate, supplemented with 10% FBS, at 37°C and 5% CO2 overnight. The following day, the cells were transfected with 12 μg of plasmid DNA per flask using lipofectamine 2000 reagent (Thermo Fisher Scientific) following the manufacturer's instructions. A reagent/ plasmid DNA ratio of 6:1 was used for transfections. At 48 h post transfection, the cells were fed selective media [DMEM supplemented with 10% FBS and 1 mg/ml G418 (Thermo Fisher Scientific), and maintained. After a week, cells growing in selective media were collected and seeded at 1 cell/well in 96-well culture plates containing selective media using Fluorescence-Activated Cell Sorting (FACS). Cells were allowed to grow and form clones at 37°C and 5% CO2 for two weeks, followed by PCSK9 Dissociation- Enhanced Lanthanide Fluorescent Immunoassay (DELFIA, reagents from PerkinElmer, Waltham, MA, USA) and western blot analysis using the media to screen for positive clones, which were expanded to establish stable cell lines.
Western blot analysis
10-15 μg/well of protein from cell lysate or liver lysate, or 20 μl/ well of media was loaded onto 4-12% BIS-TRIS gel (Thermo Fisher Scientific) and run in MOPS buffer (Thermo Fisher Scientific) for SDS-PAGE. Western blots were detected with IRDye conjugated IgGs (Licor) and scanned with a Licor Odyssey according to manufacturer’s instructions.
PCSK9 purification
PCSK9 WT and mutants were purified from the medium generated from stable HEK293 cell lines carrying Human PCSK9 WT or different mutants (lacking phenol red and containing 5% FBS) as described in Fisher et al. [21]. The purified PCSK9 was verified by western blot as well as Gelcode Blue staining (Thermo Fisher Scientific) and quantitated using a NanoDrop spectrophotometer (Thermo Fisher Scientific).
Surface plasmon resonance (Biacore)
The interaction between the LDLR ectodomain and WT or mutant PCSK9 was studied by surface plasmon resonance detection using a Biacore 3000 instrument (GE Healthcare Life Sciences, Pittsburgh, PA, USA) as described (21). Briefly, LDLR ectodomain [residues 22-788; 10 μg/ml in 10 mM sodium acetate, pH 5.0; RandD Systems, Inc (Minneapolis, MN, USA)] was coupled to a carboxymethylated dextran sensor chip (CM5, GE Healthcare Life Sciences). WT and mutant PCSK9 proteins were injected at a concentration range of 0.03-2.0 μM in a running buffer of 25 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), pH7.4, 150 mM sodium chloride, and 0.1 mM calcium chloride. The LasR ligand-binding domain was used as a control to exclude non-specific binding of PCSK9 protein to immobilized LDLR. The data was analyzed using BIAevaluation Version 4.1 software. EC50 measurements were based on two or more experiments.
LDL uptake assays
The assay was performed as described previously [21] with some modifications. HepG2 cells were seeded into poly-D-lysine coated 96-well plates (Tewksbury, MA, USA) at 30,000 cells/well in DMEM supplemented with 10% FBS (GE Healthcare Life Sciences) in 100 μl volume. After 24 h incubation at 37°C and 5% CO2 , the medium was replaced with 100 μl of DMEM lacking serum and the cells were incubated for an additional 16 h. On day 3, the medium was aspirated and 100 μl of Assay Mixture [DMEM + 10% Lipoprotein Deficient Serum (LPDS, Intracel, Frederick, MD, USA) + 10 μg/ml Alexa Fluor-546 (Thermo Fisher Scientific) labeled human LDL (Intracel) + purified PCSK9 in various concentrations] was applied to the cells. The plates were incubated at 37°C and 5% CO2 for 5 h before being washed with Tris-buffered saline (TBS, Bio-Rad, Hercules, CA, USA) and lysed with 100 μl Radioimmunoprecipitation assay (RIPA) buffer (TekNova, Hollister, CA, USA). The lysate was then transferred to a MicroFluor2 Black “U” bottom plate (Thermo Fisher Scientific). The fluorescent LDL signal was measured using an excitation wavelength of 520 nm and an emission wavelength of 580 nm. IC50 was calculated based on 2-4 replicates per point.
Cell surface LDLR expression by in-cell western
HepG2 cells were seeded into collagen coated 384-well plates (BD Biosciences) at 7,000 cells/well in DMEM supplemented with 10% FBS (GE Healthcare Life Sciences) in 25 μl volume and incubated for 24 h at 37°C and 5% CO2 , followed by an additional overnight incubation in serum-free medium. On day 3, the media was replaced with 25 μl of DMEM + 10% LPDS (Intracel) + purified PCSK9 (in various concentrations). The cells were incubated at 37°C and 5% CO2 for 5 h before fixation with 4% of paraformaldehyde (Boston BioProducts, Ashland, MA, USA). The fixed cells were blocked with blocking buffer (Licor) for 1.5 h at room temperature and probed with primary antibodies against LDLR for overnight at 4°C. The cell surface LDLR level was detected with the goat-anti-rabbit secondary antibodies conjugated with IRDye 800CW. The plate was scanned with Odyssey (Licor). DNA level stained by DRAQ5 (BioStatus, Shepshed, Leicestershire, UK) was used for well to well normalization. IC50 was calculated based on 2-4 replicates per point.
Cleavage analysis
HEK293 stable cell lines expressing wild type PCSK9 or S462P, S462A, A522T or A522G mutants were seeded into 6-well collagen coated plates (Thermo Fisher Scientific) at 1×106 cells/ well and incubated for 24 h at 37°C and 5% CO2 . The cells were treated with 20 μg/ml cycloheximide (CHX, Sigma, St. Louis, MO, USA) from 5 min to 3 h. The cells and media were collected at each time point. Cell lysate was loaded onto the SDS-PAGE and probed with anti-PCSK9 antibodies. The intensity of pro-PCSK9 and mature PCSK9 bands were analyzed with Odyssey Software (Licor). Beta-actin was used as a loading control. Secreted PCSK9 in cultured media was measured by PCSK9 DELFIA.
PCSK9 secretion assay
The secreted PCSK9 in culturing media was assessed by PCSK9 DELFIA assay modified from that previously described [23]. In brief, 96-well Immulon 4HBX plates (Thermo Fisher Scientific) were coated with 5 μg/ml of PCSK9 monoclonal antibody (mAb, E07) at 4°C overnight. 50 μl of cell culture media or PCSK9 standards were added on the plate and incubated at 37°C for 1 h. Subsequently, 50 μl of biotinylated PCSK9 mAb (B20) at 1 μg/ml in DELFIA assay buffer (Perkin Elmer) was added to the plate and incubated at RT for 1 h, followed by 50 μl of 1:1000 Streptavidin/ Europium (Perkin Elmer) incubated for 20 min at RT and finally 100 μl of DELFIA Enhance (Perkin Elmer). The plate was read on an Envision 2103 (Perkin Elmer). To account for cell line-specific differences in overexpression, PCSK9 mRNA of each mutant was measured and amounts were used to normalize the value of secreted PCSK9.
PCSK9 co-immunoprecipitation with calreticulin
Cell lysate from HEK293 stable cell lines expressing WT or S462P, S462A, A522G or A522T PCSK9 mutants was immunoprecipitated with an anti-V5 antibody, then detected with an anti-calreticulin antibody. Rabbit IgG or mouse IgG2a was incubated with the lysate as a negative control. The integrated intensity of the calreticulin band was quantitated with Odyssey Infrared Imaging System Application software. All pull downs were assessed after being normalized to the WT input.
Mouse models and animal procedures
Generation of adeno-associated virus serotype 8 (AAV8) vectors: Human PCSK9 (hPCSK9) WT cDNA was first synthesized and cloned in the pUC57 shuttle plasmid by Genscript (Piscataway, NJ, USA) with MluI and XhoI sites introduced at the 5’ and 3’ ends respectively. Site-directed mutagenesis was performed to generate two hPCSK9 mutants, S462P and A522T. The hPCSK9 WT and mutants were subcloned into MluI and XhoI sites of a pAAV cis plasmid that contains the liver-specific thyroxine binding globulin promoter (TBG). Luciferase driven by the same promoter was used as control. The AAV8 vectors were generated with the helper-free triple plasmid transfection method, and purified using double cesium chloride gradient centrifugation by the Viral Vector Core of University of Massachusetts Medical School. The titers were determined via real time PCR.
in vivo experiments: Animals were maintained in Association for Assessment and Accreditation of Laboratory Animal Care-accredited facilities at Merck Research Laboratories; all experimental procedures were approved by the Institutional Animal Care and Use Committee in conformance with the National Research Council’s Guide for the Care and Use of Laboratory Animals.
Nine to ten week old female Pcsk9-/- mice were obtained from Taconic (Rensselaer, NY, USA). Mice were housed in temperature-, humidity-, and light-controlled rooms with ad libitum access to water and diet 7012 (Research Diets, New Brunswick, NJ, USA). AAV-luciferase, AAV-hPCSK9 WT or mutant was administrated to the Pcsk9-/- mice via tail vein injection at the dose of 1×1010 genome copy per mouse in 100 μl physiological saline.
On day 14 post AAV treatment, mice were subjected to a 4 h fast, and blood was collected from tail vein to obtain plasma for PCSK9 measurements. On day 28, food was removed from the cages at 7 AM; 3 h later, the mice were then given an intraperitoneal injection of 99.8%D2O (Sigma, 20 μl/g body weight). Four hours post-D2O administration, mice were euthanized and blood was collected for subsequent plasma analysis and livers were collected for measures of mRNA, and LDLR protein level. Plasma measurements were performed for total cholesterol, LDL-c, high-density lipoprotein cholesterol (HDL-c), triglycerides (TG), hPCSK9, ApoB, glucose, insulin, aspartate aminotransferase (AST), alanine aminotransferase (ALT) and for cholesterol and fatty acid (FA) synthesis.
Analytical methods for plasma lipids and other measures: Blood was collected in ethylenediaminetetraacetic acid (EDTA)-tubes, and spun at 13,000 RPM for 10 min to obtain plasma. Plasma was transferred to a filter tube containing the dried lipase inhibitor (paraoxon, Sigma), filtered by spinning at 13,000 RPM for 2 min at room temperature. Thereafter, protease inhibitor cocktail (Sigma) was added and plasma samples were stored at 4°C for further analysis.
To generate lipoprotein profiles, plasma was fractionated by chromatography over a Superose-6 size exclusion column (GE Healthcare Life Sciences). Cholesterol levels in the column effluent were continuously measured via in-line mixture with a commercially available enzymatic colorimetric cholesterol detection reagent (Total Cholesterol E, Wako Chemicals USA, Richmond, VA, USA) followed by downstream spectrophotometric detection of the reaction products at 600 nm absorbance. The first peak of cholesterol eluted from the column was attributed to very low density lipoprotein cholesterol (VLDL-c), the second peak to LDL-c and the third to HDL-c; the area under each peak was calculated using software provided with the fast protein liquid chromatography (FPLC, GE Healthcare Life Sciences). To calculate the cholesterol concentration for each lipoprotein fraction, the ratio of the corresponding peak area to total peak area was multiplied by the total cholesterol concentration measured in the sample. Total cholesterol and TG level in plasma samples were measured separately by using Total Cholesterol E and Infinity Triglycerides kits (Thermo Fisher Scientific), respectively.
Glucose, insulin, AST, ALT were measured with standard kits from Roche Diagnostics (Indianapolis, IN, USA) on the Roche Clinical Chemistry Analyzer P modular (Roche Diagnostics) following the manufacturer’s instruction.
Cholesterol synthesis rate was measured with Liquid chromatography–mass spectrometry (LC/MS) usingD2O tracer as previously described [24]. In brief, all animals were given a bolus ofD2O after which the 2H-labeling of water, cholesterol, cholic acid and ApoA1 were measured at various time intervals. The cholesterol synthesis rate was calculated based on the mathematic model described by Previs [24].
Data analysis
Data are presented as mean ± standard error of the mean. Oneway ANOVA tests were used for statistical analysis. P-values that were <0.05 were considered statistically significant.
Results
in vitro characterization of PCSK9 mutants
Binding of WT and PCSK9 C-terminal mutants to the immobilized ectodomain of LDLR was evaluated by Biacore. In agreement with previous studies, WT PCSK9 bound LDLR with an affinity of 200 nM [22], and the gain of function (GOF) mutant, D374Y PCSK9, demonstrated an improved affinity (KD=33 nM) while the affinity of S127R PCSK9 was similar to WT. Notably, two naturally occurring non-synonymous PCSK9 polymorphisms in the C-terminal domain (S462P and A522T), and conservative mutations at these positions (S462A and A522G), all bound LDLR in a similar manner to WT PCSK9 (Table 1 and Figure 1A).
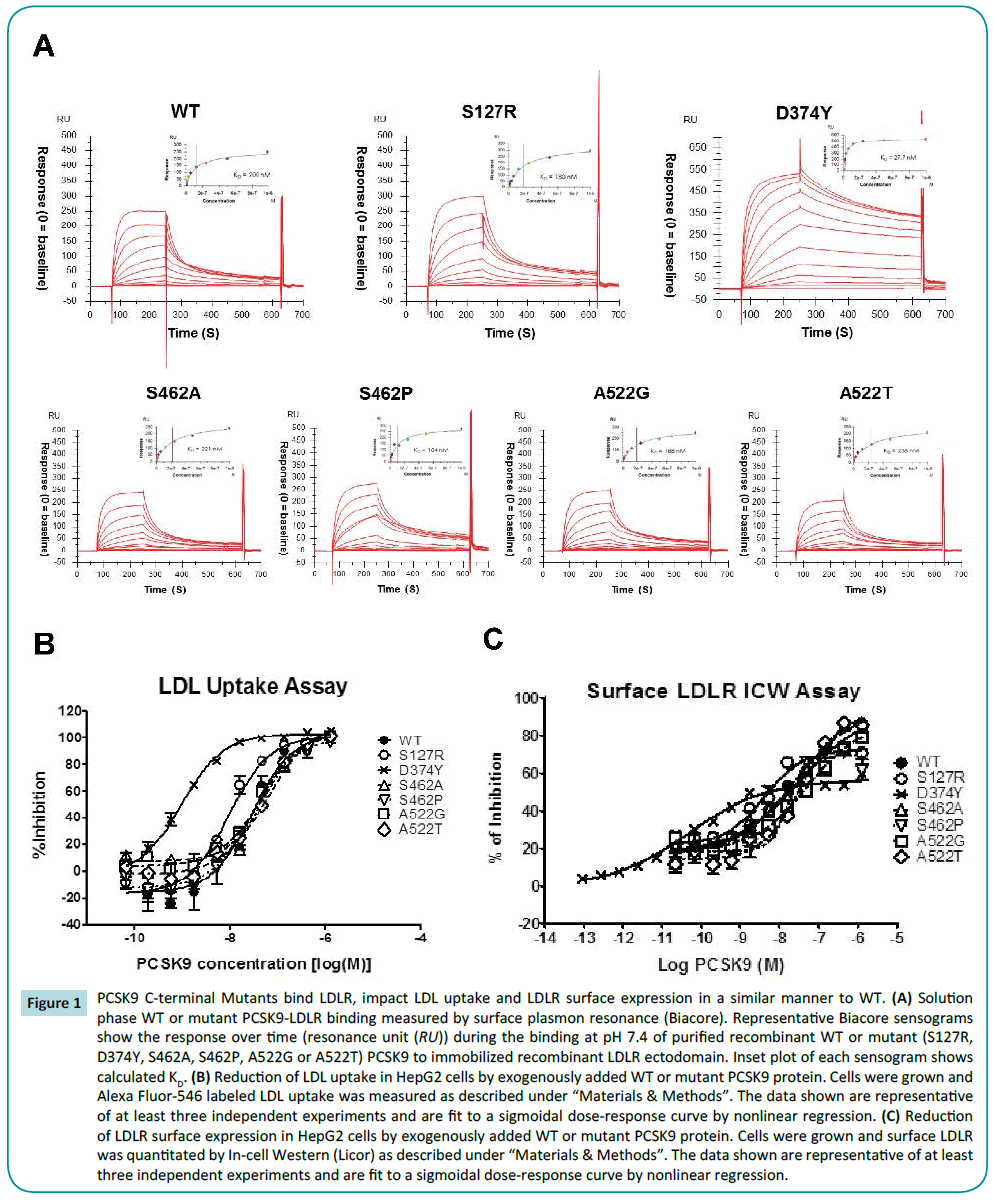
Figure 1: PCSK9 C-terminal Mutants bind LDLR, impact LDL uptake and LDLR surface expression in a similar manner to WT. (A) Solution phase WT or mutant PCSK9-LDLR binding measured by surface plasmon resonance (Biacore). Representative Biacore sensograms show the response over time (resonance unit (RU)) during the binding at pH 7.4 of purified recombinant WT or mutant (S127R, D374Y, S462A, S462P, A522G or A522T) PCSK9 to immobilized recombinant LDLR ectodomain. Inset plot of each sensogram shows calculated KD. (B) Reduction of LDL uptake in HepG2 cells by exogenously added WT or mutant PCSK9 protein. Cells were grown and Alexa Fluor-546 labeled LDL uptake was measured as described under “Materials & Methods”. The data shown are representative of at least three independent experiments and are fit to a sigmoidal dose-response curve by nonlinear regression. (C) Reduction of LDLR surface expression in HepG2 cells by exogenously added WT or mutant PCSK9 protein. Cells were grown and surface LDLR was quantitated by In-cell Western (Licor) as described under “Materials & Methods”. The data shown are representative of at least three independent experiments and are fit to a sigmoidal dose-response curve by nonlinear regression.
| Mutant |
Phenotype |
Biacore
(KD, nM) |
LDL uptake
(IC50, nM) |
Cell surface
LDLR (IC50, nM) |
| WT |
|
200 |
29.1 |
13.0 |
| D374Y |
GOF |
33 |
1.0 |
0.1 |
| S127R |
GOF |
180 |
9.4 |
4.1 |
| S462P |
LOF |
104 |
29.3 |
16.8 |
| S462A |
|
221 |
73.2 |
23.2 |
| A522T |
LOF |
188 |
48.5 |
43.2 |
| A522G |
|
238 |
43.1 |
48.6 |
Table 1: Biophysical responses and function of PCSK9 C-terminal mutants. Recombinant proteins were purified from the media of stable HEK293 cells over-expressing WT PCSK9, S462P, S462A, A522T, A522G, S127R or D374Y mutants. The binding of WT or mutant PCSK9 to immobilized recombinant LDLR ectodomain was measured by surface plasmon resonance (Biacore). The inhibition of LDL uptake and cell surface LDLR level by WT or mutant PCSK9 was measured in HepG2 cells as described under “Materials and Methods”. PCSK9 D374Y and S127R variants were used as controls.
Monitoring the ability of WT PCSK9 and mutants to modulate LDL uptake via the LDLR in HepG2 cells revealed that WT PCSK9 inhibits LDL uptake with an EC50 of 29.1 nM. While D374Y and S127R GOF PCSK9 mutants increased LDL uptake as previously demonstrated (22) (IC50 1 and 9.4 nM, respectively), S462P, S462A, A522T and A522G PCSK9 possessed similar IC50s to WT PCSK9 (IC50 29.3, 73.2, 48.5, and 43.1 nM, respectively) (Table 1 and Figure 1B). Complementary studies measured the impact of WT and mutant PCSK9 on surface expression of LDLR on HepG2 cells similarly demonstrates that the GOF PCSK9 mutants D374Y and S127R internalize LDLR with greater potency than WT PCSK9 (IC50, 0.1 and 4.1 nM, relative to 13 nM) while S462P, S462A, A522T and A522G PCSK9 possessed similar IC50 ’s to WT PCSK9 (16.8, 23.2, 43.2, and 48.6 nM, respectively) (Table 1 and Figure 1C).
Monitoring PCSK9 cleavage and secretion of WT and variants
The impact of mutation on the cleavage of PCSK9 in the endoplasmic reticulum (ER) of HEK293 cells stably expressing each variant was assessed by treating with CHX to inhibit protein synthesis followed by an additional 5 h time course. At the beginning of this semi-quantitative study, intracellular PCSK9 of WT, S462A, S462P and A522T was ~60% cleaved and ~40% proPCSK9, while S127R PCSK9 was predominantly in the proPCSK9 form (~80% uncleaved), as expected [22]. Notably, multiple studies indicated that A522G PCSK9 exhibited a somewhat intermediate phenotype with approximately ~38% cleaved upon initiation of the experiment (Figures 2A and 2B).
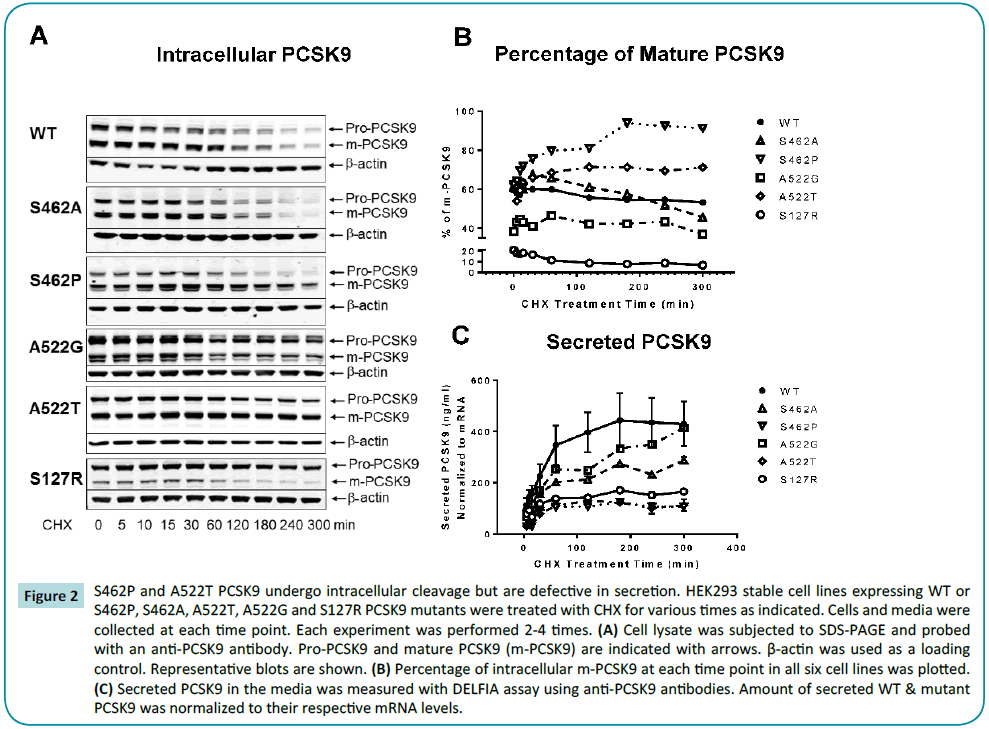
Figure 2: S462P and A522T PCSK9 undergo intracellular cleavage but are defective in secretion. HEK293 stable cell lines expressing WT or S462P, S462A, A522T, A522G and S127R PCSK9 mutants were treated with CHX for various times as indicated. Cells and media were collected at each time point. Each experiment was performed 2-4 times. (A) Cell lysate was subjected to SDS-PAGE and probed with an anti-PCSK9 antibody. Pro-PCSK9 and mature PCSK9 (m-PCSK9) are indicated with arrows. β-actin was used as a loading control. Representative blots are shown. (B) Percentage of intracellular m-PCSK9 at each time point in all six cell lines was plotted. (C) Secreted PCSK9 in the media was measured with DELFIA assay using anti-PCSK9 antibodies. Amount of secreted WT & mutant PCSK9 was normalized to their respective mRNA levels.
Over the course of the study the total percentage of mature WT PCSK9 remains quite constant (t=0, ~60% to t=5 h, 53%. In a similar manner, the relative amounts of pro/mature PCSK9 for A522G and S127R remain constant during the time-course, although S127R PCSK9 is predominantly in the proPCSK9 form (~20% mature PCSK9 at time 0 and decreased to almost none by the end of 5 h), consistent with previous reports [22]. In contrast, S462P and A522T PCSK9 increase in their percentage of mature PCSK9 to ~91% and ~71%, respectively, while S462A PCSK9 shows an initial transient increase before processing over time results in less of this species relative to WT PCSK9.
After CHX treatment, WT PCSK9 is secreted efficiently, reaching a steady state level after approximately 3 h. In contrast, as reported [22], secretion of S127R PCSK9 was reduced by approximately 80% at 5 h due to reduced cleavage to mature protein (Figures 2B and 2C). While A522G and S462A PCSK9 were secreted at levels approaching those of WT PCSK9, S462P and A522T PCSK9 exhibited significantly lower secretion (~20% of WT PCSK9) following CHX treatment.
S462P and A522T PCSK9 co-immunoprecipitate with ER stress marker, calreticulin
The observation that most of the PCSK9 mutants are cleaved successfully yet fail to be secreted efficiently indicated that they may be trapped in the ER or Golgi complex. Using an endoglycosidase H cleavage assay, Cameron et al. [19] demonstrated that S462P PCSK9 was sensitive to the enzyme; therefore the majority of the protein is likely retained in ER. Co-staining of WT or mutated PCSK9 with ER or Golgi complex markers showed that most of S462P and A522T PCSK9 are predominantly located in the ER while WT PCSK9 trafficked from ER to Golgi and is eventually secreted out over the 5 hours following CHX treatment (data not shown). To verify that S462P and A522T PCSK9 do not fold correctly, resulting in ER entrapment and subsequent degradation, lysate from all four PCSK9 variants (S462P, S462A, A522T and A522G) and WT PCSK9 was coimmunoprecipited with calreticulin, the ER stress marker (Figure 3) with all pull downs normalized to the WT input. While the conservative mutant S462A PCSK9 had a normalized pull down similar to WT PCSK9, mutant S462P PCSK9 was enriched five-fold, suggesting higher levels of S462P PCSK9: calreticulin association, consistent with an ER-localization. The normalized pull-down of A522T PCSK9 was 3.6-fold higher than WT PCSK9, indicating that it was also concentrated in the ER, although not to the degree seen with S462P PCSK9. In comparison to the more conservative mutant, A522G PCSK9, showed modest accumulation in the ER with a normalized pull down of approximately 2.5-fold higher than WT PCSK9.
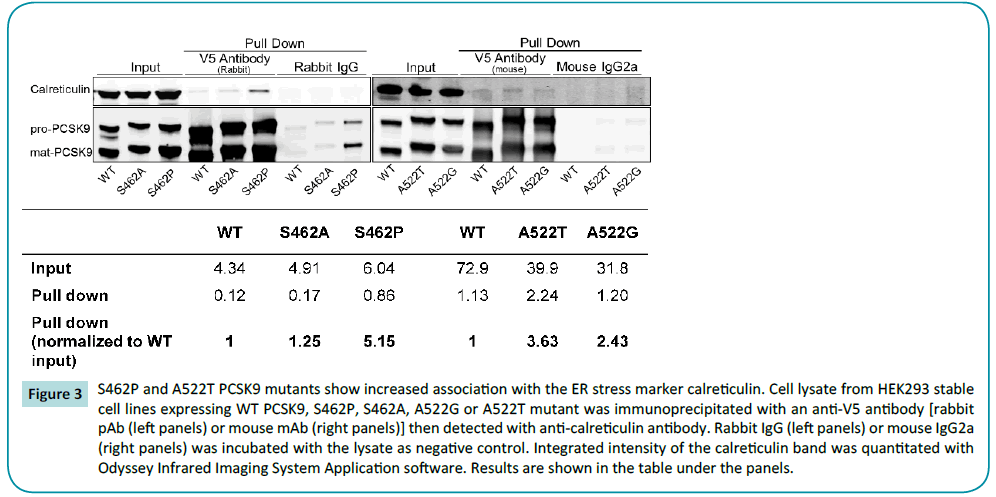
Figure 3: S462P and A522T PCSK9 mutants show increased association with the ER stress marker calreticulin. Cell lysate from HEK293 stable cell lines expressing WT PCSK9, S462P, S462A, A522G or A522T mutant was immunoprecipitated with an anti-V5 antibody [rabbit pAb (left panels) or mouse mAb (right panels)] then detected with anti-calreticulin antibody. Rabbit IgG (left panels) or mouse IgG2a (right panels) was incubated with the lysate as negative control. Integrated intensity of the calreticulin band was quantitated with Odyssey Infrared Imaging System Application software. Results are shown in the table under the panels.
S462P and A522T PCSK9 profiles in vivo
To understand whether the predicted destabilization of the C-terminal domain translates into altered PCSK9 function in vivo, the two naturally occurring, non-synonymous polymorphisms exhibiting more extensive in vitro differentiation versus WT (S462P and A522T) were expressed in Pcsk9-/- mice. In these studies, female Pcsk9-/- or control mice were fed normal chow diet until 9-10 weeks old. Thereafter, control or WT/mutant hPCSK9- AAV was delivered to mice at the start of the experiment, and 4 weeks later animals were profiled (Figure 4A).
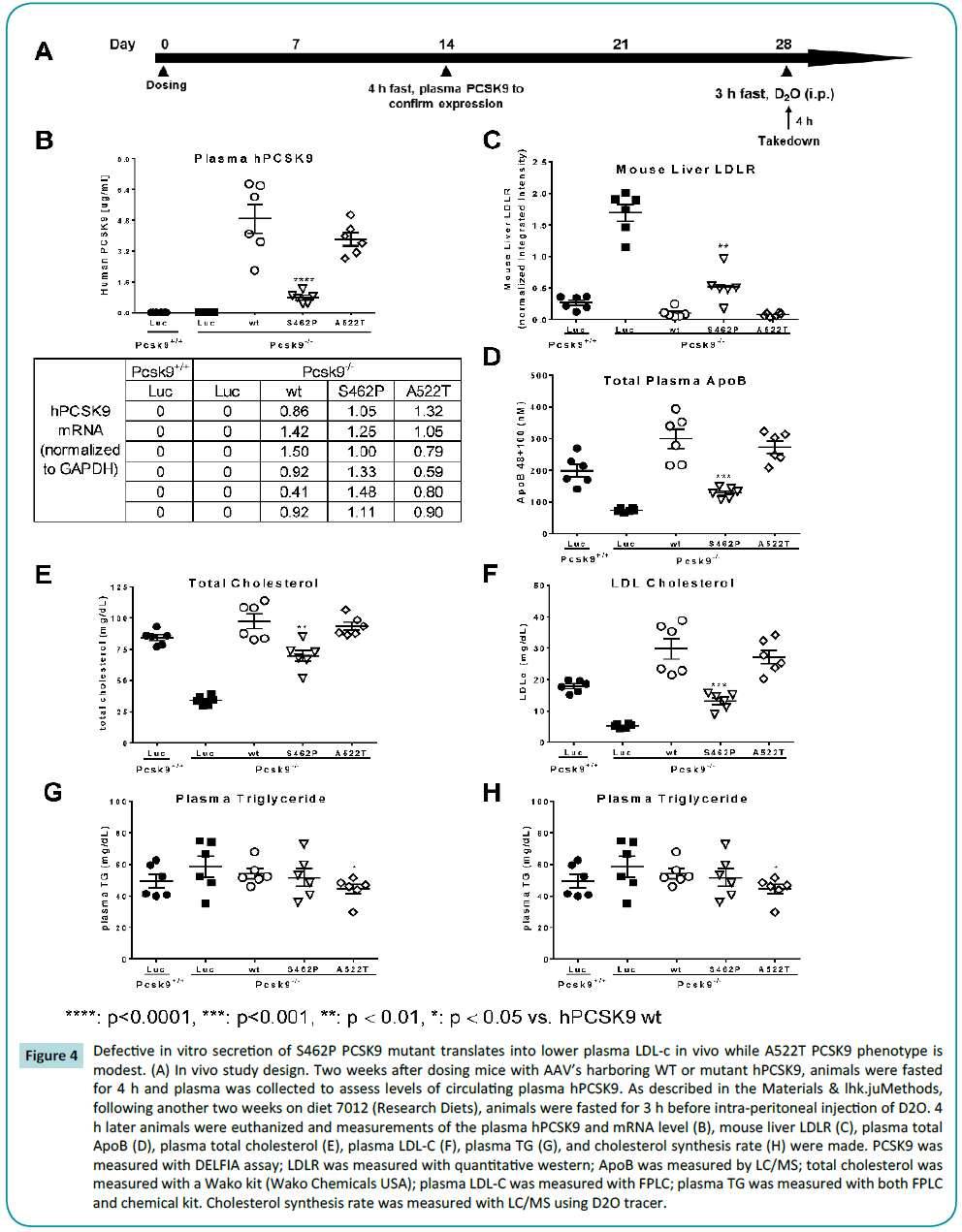
Figure 4: Defective in vitro secretion of S462P PCSK9 mutant translates into lower plasma LDL-c in vivo while A522T PCSK9 phenotype is modest. (A) In vivo study design. Two weeks after dosing mice with AAV’s harboring WT or mutant hPCSK9, animals were fasted for 4 h and plasma was collected to assess levels of circulating plasma hPCSK9. As described in the Materials & lhk.juMethods, following another two weeks on diet 7012 (Research Diets), animals were fasted for 3 h before intra-peritoneal injection of D2O. 4 h later animals were euthanized and measurements of the plasma hPCSK9 and mRNA level (B), mouse liver LDLR (C), plasma total ApoB (D), plasma total cholesterol (E), plasma LDL-C (F), plasma TG (G), and cholesterol synthesis rate (H) were made. PCSK9 was measured with DELFIA assay; LDLR was measured with quantitative western; ApoB was measured by LC/MS; total cholesterol was measured with a Wako kit (Wako Chemicals USA); plasma LDL-C was measured with FPLC; plasma TG was measured with both FPLC and chemical kit. Cholesterol synthesis rate was measured with LC/MS using D2O tracer.
Plasma levels of hPCSK9 were measured in all the mice (Figure 4B). hPCSK9 was undetectable in the WT Pcsk9+/+ mouse control and the Pcsk9-/- mouse. In the three experimental groups of mice where human WT, S462P or A522T PCSK9 was inserted, hPCSK9 was found at comparably high levels in the WT and A522T PCSK9 variants (~5.0 and 4.0 μg/ml, respectively) and at significantly reduced levels (~1 μg/ml) in S462P PCSK9. These levels are all elevated relative to that normally observed in C57BL/6 background mice (routinely ~150-200 ng/ml). Notably, liver PCSK9 mRNA transcript levels were comparable across all groups.
Mouse liver LDLR expression levels (Figure 4C) were elevated in Pcsk9-/- mice relative to WT controls (Pcsk9+/+). As expected, expression of WT hPCSK9 in the Pcsk9-/- mice led to a reduction in liver LDLR levels. While A522T PCSK9 reduced LDLR expression in a similar manner to WT PCSK9 in the Pcsk9-/- mouse background, S462P PCSK9 was less efficient in the abrogation of LDLR expression. In a reciprocal manner, ApoB, LDL-c and total cholesterol levels were decreased in Pcsk9-/- mice relative to the WT Pcsk9+/+ mice. Further, levels of all three were increased relative to Pcsk9-/- mice following expression of WT, S462P and A522T PCSK9, although this effect was significantly more modest in the S462P PCSK9 mice (Figures 4D, 4E and 4F).
Notably, in this study we also evaluated impact on plasma TG and the rate of cholesterol synthesis. With the exception of a modest effect on TG’s with A522T PCSK9, neither of these parameters were altered by the manipulations described (Figures 4G and 4H). Likewise, Glucose, insulin, liver ALT and AST were unchanged in any group (Supplement Figure 1).
Molecular modelling
To gain insight into the mechanistic basis of non-synonymous polymorphisms in hPCSK9 (S462P and A522T PCSK9), we employed the Protein Design module in Discovery Studio (Accelrys, San Diego, CA, USA) to assess molecular stability. Using Protein database coordinates for WT [PDB code 3bps] [25], and mutant [S462P and A522T PCSK9] (Figure 5A), stability of the mutants was assessed as interaction energy differences (Figure 5B). While S462A PCSK9 had a similar energetic calculation as WT PCSK9, a slight destabilizing effect was calculated for A522G and A522T PCSK9 with a highly significant, ~30 kcal/mol and destabilization energy for S462P PCSK9.
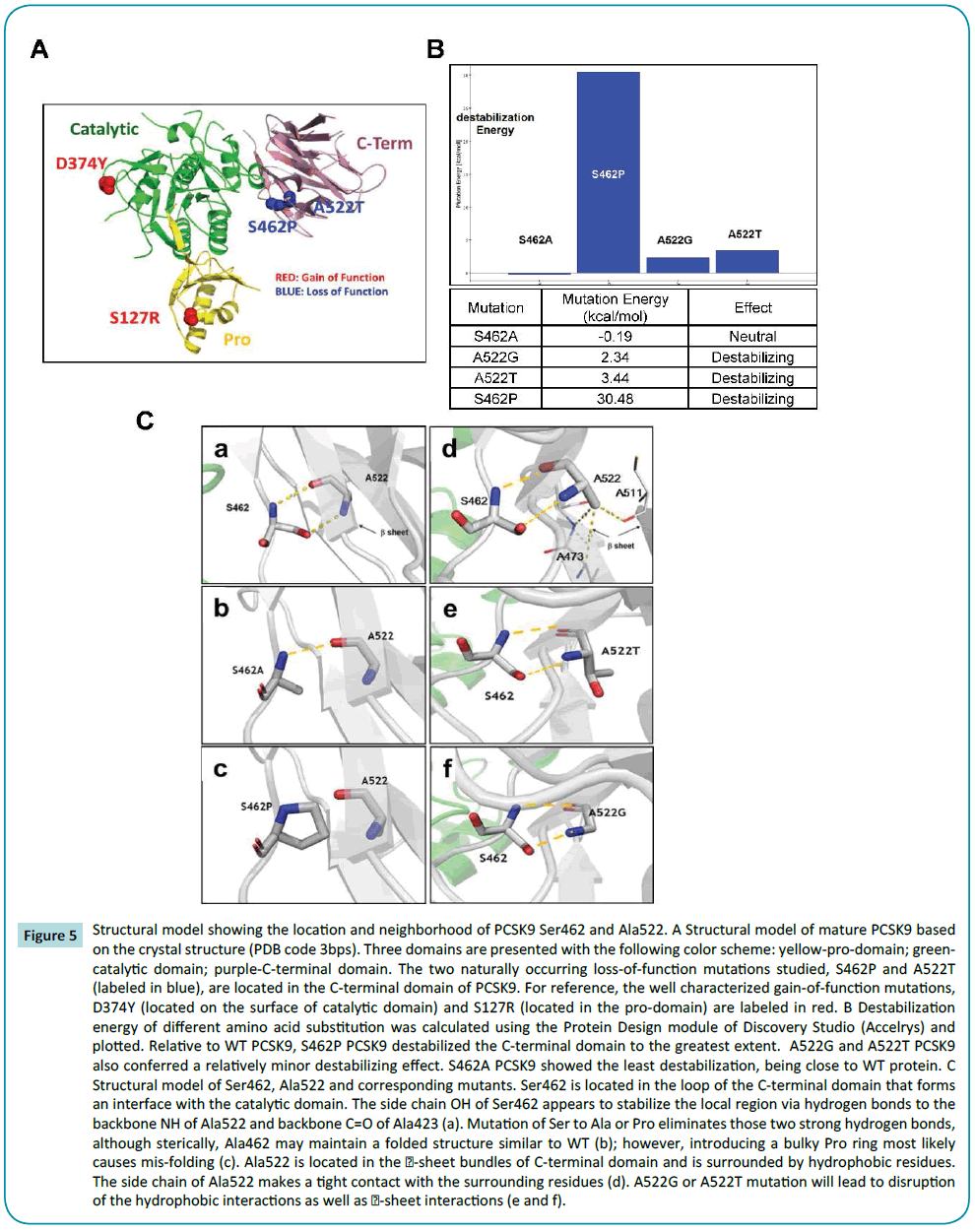
Figure 5: Structural model showing the location and neighborhood of PCSK9 Ser462 and Ala522. A Structural model of mature PCSK9 based on the crystal structure (PDB code 3bps). Three domains are presented with the following color scheme: yellow-pro-domain; greencatalytic domain; purple-C-terminal domain. The two naturally occurring loss-of-function mutations studied, S462P and A522T (labeled in blue), are located in the C-terminal domain of PCSK9. For reference, the well characterized gain-of-function mutations, D374Y (located on the surface of catalytic domain) and S127R (located in the pro-domain) are labeled in red. B Destabilization energy of different amino acid substitution was calculated using the Protein Design module of Discovery Studio (Accelrys) and plotted. Relative to WT PCSK9, S462P PCSK9 destabilized the C-terminal domain to the greatest extent. A522G and A522T PCSK9 also conferred a relatively minor destabilizing effect. S462A PCSK9 showed the least destabilization, being close to WT protein. C Structural model of Ser462, Ala522 and corresponding mutants. Ser462 is located in the loop of the C-terminal domain that forms an interface with the catalytic domain. The side chain OH of Ser462 appears to stabilize the local region via hydrogen bonds to the backbone NH of Ala522 and backbone C=O of Ala423 (a). Mutation of Ser to Ala or Pro eliminates those two strong hydrogen bonds, although sterically, Ala462 may maintain a folded structure similar to WT (b); however, introducing a bulky Pro ring most likely causes mis-folding (c). Ala522 is located in the ï¢-sheet bundles of C-terminal domain and is surrounded by hydrophobic residues. The side chain of Ala522 makes a tight contact with the surrounding residues (d). A522G or A522T mutation will lead to disruption of the hydrophobic interactions as well as sheet interactions (e and f).
Location of the mutations and their spatial relationship is shown in Figures 5A and 5C. Ser462 is located in the loop of the C-terminal domain that interfaces with the catalytic domain. The side chain OH of Ser462 appears to stabilize the local region via hydrogen bonds to the backbone NH of Ala522 and backbone C=O of Ala522 (Figure 5C-a). Mutant S462P PCSK9 has a strong impact on destabilization of this region. Ala522 is part of the β6 sheet and interacts with Ser462 via two hydrogen bonds as shown in Figure 5C. The energetics indicated that mutant S462A has the least disruption. Although losing one hydrogen bond, the mutation seems to compensate by stabilizing the region by releasing the possible tight interactions. Sterically, Ala462 may maintain the folding structure (Figure 5C-b). However, mutation of Ser462 to Pro would not only lose two hydrogen bonds, the Pro ring would also have an unwanted contact with the β sheet where Ala522 is located, likely causing destabilization (Figure 5Cc).
Ala522 is located in the β-sheet bundles of the C-terminal domain. Ala522 is surrounded by hydrophobic residues. The side chain of Ala522 makes a tight contact with the surrounding residues in other β-sheets (Figure 5C-d). A522G or A522T PCSK9 mutations will lead to the disruption of the hydrophobic interactions and destabilization of the β-sheet interaction (Figures 5C-e and 5C-f).
Discussion
In the current study, we profiled human non-synonymous polymorphisms in the C-terminal domain of PCSK9 (S462P and A522T) utilizing in vitro and in vivo approaches. Two conservative mutants, S462A and A522G, were made in parallel to facilitate the exploration of the impact of these two sites. The rational of replacing Ser to Ala is to eliminate the hydrogen bond between Ser462 side chain -OH and backbone -NH of A522. Since Ala would not introduce the bulky side chain as Pro does, an unwanted steric interaction would not be added. Likewise, substitution of Ala522 with Gly reduces hydrophobic interactions of the residue with Ala473 and Ala511.
in vitro, S462P and A522T versions of PCSK9 auto-cleaved in a similar manner to WT PCSK9, however, secretion was less efficient, likely due to mis-folding, as suggested by an increased association with calreticulin. in vivo, despite hPCSK9 equivalent transcript levels in mouse liver, plasma hPCSK9 levels were moderately/significantly lower for A522T and S462P respectively, correlating with the magnitude of reduced plasma cholesterol levels. Molecular modeling indicates that S462P and A522T mutations destabilize the C-terminal domain of PCSK9 to varying degrees with the severity correlating with the impact on in vitro folding/secretion and in vivo phenotype. These observations extend those of Cameron et al. [19] with regard to S462P PCSK9, and provide insight into the phenotype of A522T PCSK9.
To date, human polymorphisms in PCSK9 have been found in all three domains [19,22,26-30]. Generally, mutations in the prodomain or catalytic domain either directly affect the autocatalytic activity of PCSK9 (S217R [22,27], D129G [27], N354I [26]), thus reducing protein secretion and/or alter the affinity of PCSK9 binding to LDLR (R46L [29], D374Y [22], R215H [26]). With respect to the C-terminal domain of PCSK9, previous reports [14,15,17,18] have demonstrated that it is not required for LDLR binding, but rather for its trafficking and degradation. Furthermore, Holla et al. have revealed the role of this domain in PCSK9 secretion, or LDLR internalization, through mutating conserved amino acids in this domain [11]. Here we find that the LOF mutants, S462P and A522T PCSK9, interact with LDLR normally as judged by multiple binding approaches and LDL uptake, indicating that the LOF phenotype exhibited by these mutants cannot be accounted for by the interaction between PCSK9 and LDLR and, more likely, results from improper PCSK9 processing.
Consistent with the concept of improper processing/ secretion, Cameron et al. [19] demonstrated that HEK293 cells overexpressing S462P PCSK9 exhibited normal auto-cleavage but reduced protein secretion. In agreement with Cameron et al., we found that although S462P and A522T PCSK9 autocleave appropriately, secretion over 5 h was approximately one quarter that of WT PCSK9. To assess why these variants of PCSK9 exhibited reduced secretion we evaluated their interaction with the ER stress marker calreticulin, which interacts with misfolded proteins and targets them for degradation. While both A522T and S462P PCSK9 showed increased interaction with calreticulin, the association was significantly greater for the S462P polymorphism. Notably, while Holla’s scanning mutagenesis studies suggested that interactions between the C-terminal and catalytic domains of PCSK9 are mediated by electrostatic interactions [11], the nonsynonymous polymorphisms characterized in the current study suggest that changes in C-terminal domain bulk may impact folding and possibly inter-domain interactions.
Despite comparable levels of PCSK9 transcript in the liver of mice transduced with WT and S462P/A522T PCSK9-AAV’s, the plasma level of hPCSK9 in AAV8-S462P treated mice was significantly lower than that of those treated with AAV8-WT. As the consequence of the reduced plasma S462P hPCSK9 levels, AAV8- S462P treated mice demonstrated a significantly lower level of plasma total cholesterol, LDL-c and ApoB along with higher levels of liver LDLR. In contrast, and in line with the in vitro phenotyping, AAV8-A522T treated mice exhibited only slightly decreased total cholesterol and LDL-c levels consistent with the trend towards lower plasma A522T hPCSK9 levels relative to AAV8-WT treated mice. Consistent with these observations, while molecular modeling indicates that the S462P polymorphism leads to a significant de-stabilization of the C-terminal domain, the A522T PCSK9 polymorphism is more subtle (~10% of the de-stabilization predicted for S462P).
The time required for the LDLR to complete a cycle of lipoprotein internalization has been reported to be approximately 10 min [31,32]. We have observed the same degree of alteration on hepatic cell (HepG2) surface LDLR concentration after 5 h treatment of the cells with the purified WT PCSK9 or mutants, which indicates that there is no impact on the recycling of LDLR after its internalization in such an acute setting. However, studies in more chronic conditions may provide further insight. The observation that mice with S462P or A522T PCSK9 had normal liver LDLR levels proportional to circulating PCSK9 suggests that there is no impact on receptor cycling, with the caveat that the hPCSK9 expression level of these mutants was much greater than normally seen in a mouse and thus, may not provide the granularity of information that we would hope to have to answer this particular question.
Presentation of individuals bearing S462P or A522T PCSK9 nonsynonymous polymorphisms is rare, making it challenging to understand the relative impact of these mutations on whole body lipid management. Along these lines, the single individual identified carrying the A522T PCSK9 mutation exhibits a hypocholesterolemic phenotype [19,20]. Seemingly in contrast, the individual carrying the S462P PCSK9 polymorphism had elevated cholesterol levels, however, her father, from whom she had inherited the mutation, was hypocholesterolemic. Hence, given these complexities with interpreting underpowered human data, here we exploit the consistency of the mouse genetic background to examine the relative impact of these polymorphisms. The relative in vivo severity of the S462P polymorphism is highly consistent with molecular modeling and in vitro phenotyping studies, furthering the argument for a role of the C-terminal domain of PCSK9 in protein secretion.
Conflict of Interest
The authors are employees of Merck and Co., Inc.
No conflicts of interest as such.
References
- Horton JD, Cohen JC, Hobbs HH (2009) PCSK9: A convertasethat coordinates LDL catabolism. J Lipid Res 50: S172-S177
- Peterson AS, Fong LG, Young SG (2008) PCSK9 function and physiology. J Lipid Res 49: 1152-1156
- Horton JD, Cohen JC, Hobbs HH (2007) Molecular biology of PCSK9: Its role in LDL metabolism. Trends BiochemSci32: 71-77.
- Kotowski IK, Pertsemlidis A, Luke A, Cooper RS, Vega GL, et al. (2006)A spectrum of PCSK9 alleles contributes to plasma levels of low-density lipoprotein cholesterol. Am J Hum Genet 78: 410-422.
- Cohen JC,Boerwinkle E, Mosley TH Jr, Hobbs HH (2006) Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med 354: 1264-1272.
- Victor RG, Haley RW, Willett DL, Peshock RM, Vaeth PC, et al.(2004) The Dallas heart study: A population-based probability sample for the multidisciplinary study of ethnic differences in cardiovascular health. Am J Cardiol93: 1473-1480.
- Giunzioni I, Tavori H (2006) New developments in atherosclerosis: Clinical potential of PCSK9 inhibition. Vasc Health Risk Manag 11: 493-501.
- Markham A(2015) Alirocumab: First global approval. Drugs 75: 1699-1705.
- Markham A(2015)Evolocumab: First global approval. Drugs 75: 1567-1573.
- Bottomley MJ, Cirillo A,Orsatti L, Ruggeri L, Fisher TS, et al. (2009) Structural and biochemical characterization of the wild type PCSK9-EGF(AB) complex and natural familial hypercholesterolemia mutants. J BiolChem 284: 1313-1323.
- Holla ÃYL, Cameron J, Tveten K, Strom TB, Berge KE, et al.(2011) Role of the C-terminal domain of PCSK9 in degradation of the LDL receptors. J Lipid Res 52: 1787-1794
- Leren TP(2014) Sorting an LDL receptor with bound PCSK9 to intracellular degradation. Atherosclerosis 237: 76-81.
- Schiele F, Park J, Redemann N,Luippold G, Nar H(2014) An antibody against the C-terminal domain of PCSK9 lowers LDL cholesterol levels in vivo. J MolBiol 426: 843-852.
- Tveten K, Holla OL, Cameron J, Strom TB, Berge KE, et al. (2012) Interaction between the ligand-binding domain of the LDL receptor and the C-terminal domain of PCSK9 is required for PCSK9 to remain bound to the LDL receptor during endosomal acidification. Hum Mol Genet 21: 1402-1409.
- Zhang DW,Garuti R, Tang WJ, Cohen JC, Hobbs HH (2008) Structural requirements for PCSK9-mediated degradation of the low-density lipoprotein receptor. P NatlAcadSci USA 105: 13045-13050.
- Han B, Eacho PI, Knierman MD, Troutt JS, Konrad RJ, et al. (2014) Isolation and characterization of the circulating truncated form of PCSK9. J Lipid Res 55: 1505-1514.
- Saavedra YG, Day R, Seidah NG (2012) The M2 module of the Cys-His-rich domain (CHRD) of PCSK9 protein is needed for the extracellular low-density lipoprotein receptor (LDLR) degradation pathway. J BiolChem 287: 43492-43501.
- Poirier S, Hamouda HA, Villeneuve L, Demers A, Mayer G(2016) Trafficking dynamics of PCSK9-induced LDLR degradation: Focus on human PCSK9 mutations and C-terminal domain. PloS one 11: e0157230.
- Cameron J,Holla OL, Laerdahl JK, Kulseth MA, Berge KE, et al. (2009) Mutation S462P in the PCSK9 gene reduces secretion of mutant PCSK9 without affecting the autocatalytic cleavage. Atherosclerosis 203: 161-165.
- Fasano T, Cefalu AB, Di Leo E, Noto D, Pollaccia D,et al.(2007) A novel loss of function mutation of PCSK9 gene in white subjects with low-plasma low-density lipoprotein cholesterol. ArteriosclThrom Vas 27: 677-681.
- Fisher TS, Lo Surdo P,Pandit S,Mattu M, Santoro JC, et al. (2007) Effects of pH and low density lipoprotein (LDL) on PCSK9-dependent LDL receptor regulation. J BiolChem 282: 20502-20512.
- Pandit S, Wisniewski D, Santoro JC, Ha S, Ramakrishnan V, et al.(2008) Functional analysis of sites within PCSK9 responsible for hypercholesterolemia. J Lipid Res 49: 1333-1343
- Ni YG, Condra JH, Orsatti L, Shen X, Di Marco S, et al. (2010) A proproteinconvertasesubtilisin-like/kexin type 9 (PCSK9) C-terminal domain antibody antigen-binding fragment inhibits PCSK9 internalization and restores low density lipoprotein uptake. J BiolChem 285: 12882-12891.
- Previs SF, McLaren DG, Wang SP, Stout SJ, Zhou H, et al. (2014) New methodologies for studying lipid synthesis and turnover: looking backwards to enable moving forwards. BiochimBiophysActa1842: 402-413.
- Kwon HJ, Lagace TA, McNutt MC, Horton JD, Deisenhofer J (2008) Molecular basis for LDL receptor recognition by PCSK9. ProcNatlAcadSci USA 105: 1820-1825.
- Cameron J, Holla OL, Laerdahl JK, Kulseth MA, Ranheim T, et al. (2008) Characterization of novel mutations in the catalytic domain of the PCSK9 gene. J Intern Med 263: 420-431.
- Homer V, Marais D, Laurie A, Charlton F, Sullivan D, et al. (2007)Identification and molecular characterization of new PCSK9 missense mutations associated with familial hypercholesterolemia. ArteriosclThrom Vas 27: E122-E122.
- Norata GD, Garlaschelli K, Grigore L,Raselli S, Tramontana S, et al. (2010) Effects of PCSK9 variants on common carotid artery intima media thickness and relation to ApoE alleles. Atherosclerosis 208: 177-182.
- Strom TB, Holla OL, Cameron J, Berge KE, Leren TP(2010) Loss-of-function mutation R46L in the PCSK9 gene has little impact on the levels of total serum cholesterol in familial hypercholesterolemia heterozygotes. ClinChimActa 411: 229-233.
- Sun XM, Eden ER, Tosi I, Neuwirth CK, Wile D, et al. (2005) Evidence for effect of mutant PCSK9 on apolipoprotein B secretion as the cause of unusually severe dominant hypercholesterolaemia. Hum Mol Genet 14: 1161-1169.
- Cunningham D, Danley DE, Geoghegan KF, Griffor MC, Hawkins JL, et al. (2007) Structural and biophysical studies of PCSK9 and its mutants linked to familial hypercholesterolemia. Nat StructMolBiol 14: 413-419.
- Basu SK, Goldstein JL, Anderson RG, Brown MS (1981)Monensin interrupts the recycling of low density lipoprotein receptors in human fibroblasts. Cell 24: 493-502.

