Aisling McMahon1*, Eimhin Dunne1,Pamela Evans2, Raphael B Merriman3 and Alistair Nichol1,4-6
Department of Anaesthesia, Intensive Care and Pain Medicine, St Vincent’s University Hospital, Elm Park, Dublin 4, Ireland
Department of Haematology, St. Vincent’s University Hospital, Elm Park, Dublin 4, Ireland
National Liver Transplant Unit, Department of Medicine, St Vincent’s University Hospital, Elm Park, Dublin 4, Ireland
Department of Intensive Care Medicine, Alfred Hospital Melbourne, Australia
Australian and New Zealand Intensive Care Research Centre, Monash University, Melbourne, Australia
School of Medicine and Medical Sciences, University College Dublin, Dublin, Ireland
Corresponding Author:
Aisling Mc Mahon
Department of Anaesthesia, Intensive Care and Pain Medicine
St Vincent’s University Hospital, Elm Park, Dublin 4, Ireland
Tel: +353861091104
E-mail: mcmahoma@tcd.ie
Received date: January 14, 2016; Accepted date: February 08, 2016; Published date: February 15, 2016
Citation: McMahon A, Dunne E, Evans P, et al. Consumptive Coagulopathy after Commencement of Continuous Renal- Replacement Therapy in Patients with Decompensated Cirrhosis: A Case Series. J Intensive & Crit Care 2016, 2:1.
Keywords
Decompensated cirrhosis; Continuous renal-replacement therapy; Disseminated intravascular coagulation; Refractory ascites; Hyponatraemia
Introduction
Cirrhosis is a significant cause of morbidity and mortality and is thought to affect up to 0.1% of the European population [1]. It accounts for 1.8% of deaths in Europe [1] and is predicted to be the 12th leading cause of death worldwide by 2020 [2]. Of particular concern is the increasing incidence of cirrhosis in the UK and Ireland [1].
The common manifestations of decompensated cirrhosis include ascites, variceal bleeding, encephalopathy and synthetic dysfunction [3]. Disordered haemostasis is a major and frequent consequence of hepatic synthetic dysfunction reflecting insufficient production of proteins that both participate in and regulate the normally finely balanced coagulation and fibrinolytic pathways. Traditionally, patients with decompensated cirrhosis were considered hypocoagulable and therefore prone to bleeding but this paradigm has recently been challenged [4]. A more nuanced interpretation would suggest that pro- and anticoagulant factors are reset in a tenuous but albeit balanced state [4,5]. Anaemia often develops secondary to gastrointestinal bleeding or hypersplenism. Thrombocytopenia also occurs as a result of decreased liver thrombopoietin secretion and splenic sequestration and fibrinolysis can also be a feature [6]. A major challenge to the management of the coagulopathy of liver disease is the absence of established and agreed upon laboratory tests that measure the disorders of haemostasis.
More recently the role of the endothelium has been considered in contributing to an increased health risk in patients with decompensated cirrhosis [7,8]. It has been proposed that the altered liver function with cirrhosis results in endothelial dysfunction. This is characterized by a reduction in the availability of nitric oxide and modifications to the proinflammatory and procoagulant activity of the endothelium. This effect can be assessed by measuring brachial artery flow mediated vasodilation (FMD). In hepatitis C virus (HCV) infection, it has been shown that the degree endothelial dysfunction, as expressed by the FMD, progresses with the degree of liver fibrosis [8]. This association may explain some of the increased morbidity and mortality in this patient cohort.
The ascites, hyponatraemia and hepatorenal syndrome that accompany decompensated cirrhosis arise from a common mechanism and reflect the increasing severity of the disordered hemodynamic consequences of cirrhosis and portal hypertension [9]. Refractory ascites occurs in up to 10% of patients with decompensated cirrhosis [10] and is commonly associated with hyponatraemia. This has led to sodium being incorporated into some scoring systems that reflect the severity of liver disease and assigning waiting-list priority for liver transplantation such as Model for End-Stage Liver Disease-Na (MELD-Na) and the United Kingdom Model for End-Stage Liver Disease (UKELD) [11].
A 2007 UK trial concluded that the 90-day mortality was increased in those with serum sodium less than 130 Meq/L and emphasized correcting sodium levels pretransplantation [12]. However a 2009 US trial found no influence on post-transplant mortality associated with pre-transplant sodium [13]. Despite these divergent signals it remains common practice to initiate continuous renal replacement therapy (CRRT) for correction of severe hyponatraemia [9] in patients with decompensated cirrhosis.
We discuss three consecutive cases when CRRT was used to manage fluid overload and/or hyponatraemia in decompensated cirrhosis resulting in severe coagulopathy, and explore potential pathophysiologic mechanisms.
Case 1
A 37-year-old female with HCV and alcohol-related decompensated cirrhosis awaiting orthotopic liver transplantation (OLT), was referred to the intensive care unit (ICU) for CRRT for management of diuretic-resistant ascites and fluid overload. The patient had a pre-existing stable coagulopathy without overt bleeding (see Table 1). The patient was receiving intravenous antibiotics for a lower limb cellulitis at the time of referral. On admission, central venous access was established uneventfully and CRRT was commenced without local or systemic anticoagulant.
One hour after initiation of CRRT, scattered petechiae were noted over her face and chest. This was accompanied by a new derangement in the coagulation profile. Over the next 24 hours, clinically significant bleeding and further coagulopathy developed (see Table 1).
Twelve hours following ICU admission, a thromboelastogram (TEG) was performed (see Figure 1). Based on this, blood products were transfused (as outlined in Table 1 at 12 hours). Results of coagulation screens performed at 12 and 18 hours became available.
Blood products were transfused to target haemoglobin above 9 g/dL and fibrinogen above 1 mg/dL. Platelets and pooled plasma (Octaplas®) were to be given as required when clinically significant bleeding occurred. These targets, combined with clinical assessment guided transfusion therapy over the following 4 days and are detailed in Supplementary Table E1. Bleeding from the central venous catheter site and oral cavity began to ease at 36 hours after starting CRRT. Coagulation screen returned to baseline at 48 hrs and CRRT was discontinued after adequate fluid removal and clinical improvement on day 4.
The patient had two further ICU admissions during her inpatient course, both with electrolyte disturbances and renal failure requiring CRRT. A worsening coagulopathy with oozing from line insertion sites occurred on both these occasions. Over the course of the second admission a total of 8 g fibrinogen, 1 pool platelets, 4 pools of Octaplas® and 5 units red cells were transfused. During the third admission, up to the time of transplantation she received 1 pool of platelets, 4g fibrinogen, 3 pools of Octaplas® and 3 units of red cells with the worst coagulation profile on these admissions showing a platelet count of 18 × 103 μL, PT 42 seconds and APTT 79.8 seconds. Liver transplantation was performed during the final admission, after which symptoms and coagulopathy resolved. The CRRT was discontinued day 3 post transplantation.
Case 2
A 55-year-old lady with decompensated cirrhosis secondary to alcoholic liver disease, being assessed for liver transplantation was admitted to the ICU requiring CRRT for management of fluid overload and pulmonary oedema. She also had a pre-existing but stable coagulopathy. On admission, blood products were prophylactically administered to facilitate central venous access (see Table 2) and CRRT was commenced without anticoagulant. Twelve hours after commencing CRRT the patient developed a coagulopathy with clinically evident bleeding as outlined in Table 2.
Factor assays were performed and showed a mainly haemostatic picture. This confirmed that a clotting factor deficiency was not the cause of the bleeding diathesis. Therefore a TEG was performed to establish if hyperfibrinolysis was present but was initially unrecordable (see Figure 2). The TEG was repeated with complete normalization of both the TEG (see Figure 3) and more importantly, a rapid clinical improvement in bleeding affirming that hyperfibrinolysis was indeed the likely dominant driver of the coagulopathy.
| Time |
0 hour |
6 hours |
12 hours |
24 hours |
| Clinically evident bleeding |
Nil |
Petechiae over face and chest |
Frank blood orally and from vascath insertion site |
|
| Laboratory results |
Hb 7.6g/dL
PT 24.5s
APTT 57.8s
Platelets 27×103µL
Na+ 136mmol/L |
Hb 7.6g/dL
PT 28.3s
APTT 102s
Platelets 22×103µL
Na+136mmol/L |
Hb 7.4g/dL
PT 60s
APTT 161s
Platelets 27 × 103µL
Fibrinogen <0.5gL |
Hb 7.9g/dL
PT 22.6s
APTT 67.8s
Platelets35×103µL
Fibrinogen 0.56g
Na+ 138mmol/L |
| Treatment |
|
1 pool platelets |
1 unit red cells
1 pool platelets
2 units Octaplas®
4g fibrinogen |
1 unit red cells
8g fibrinogen
1000iuoctaplex®
Tranexamic acid mouthwash
Parenteral vitamin K 10mg |
Table 1: Clinically evident bleeding compared to laboratory results and the treatments administered over time in hours from admission case 1.
| Time |
0 hour |
6 hours |
12 hours |
24 hours |
| Clinically evident bleeding |
|
|
Fresh ooze from central access sites |
Continuous ooze from access sites and mucosal bleeding |
| Laboratory results |
Hb 7.6g/dL
PT 33.8s
APTTnot measured
Platelets 40 ×103µL
Fibrinogen 1.26gL
Na+ 127mmol/L |
Hb 7.6g/dL
PT 24.7s
APTT 66.3s
Platelets 58×103µL
Fibrinogen 0.97gL
Na+ 125mmol/L |
Hb 8.8g/dL
PT 30.1s
APTT 120s
Platelets 41 ×103µL
Fibrinogen 0.5gL
Na+129mmol/L |
Hb 8.6g/dL
PT 22.1s
APTT 116s
Platelets 33×103µL
Fibrinogen 0.89gL
Na+ 132mmol/L |
| Treatment |
2 poolsOctaplex®
4g fibrinogen
1 pool platelets |
2g fibrinogen
I unit red cells |
Surgicel®, adrenaline and tranexamic acid soaked gauze at line sites
Tranexamic acid mouth wash |
8g fibrinogen
4 poolsOctaplas®
1400IUOctaplex®
2 pools platelets |
Table 2: Clinically evident bleeding compared to laboratory results and the treatments administered over time in hours from admission case 2.
The haemoglobin was maintained above 9 g/dL and fibrinogen levels above 1 mg/dL, with prothrombin complex concentrate (Octaplex®), cryoprecipitate, Factor VIIa and tranexamic acid as required for clinically significant bleeding.
Additional products transfused from day 1 to 4 are detailed in Supplementary Table E2. At 48 hours after commencement of CRRT, central venous catheter oozing had eased and was minimal at 54 hours. By this stage the consumptive coagulopathy had stabilised and clotting parameters returned to near baseline. One episode of melaena then occurred requiring replacement of fibrinogen and platelets and initiation of terlipressin to reduce splanchnic venous pressure. After day 5, CRRT was discontinued. Blood products were not administered unless deemed clinically necessary. During the rest of the ICU course, the patient reestablished a baseline coagulopathy with an INR of 2-3.8, platelets of 35-50 × 103 μL, APTT 60-70 seconds and fibrinogen < 0.5 mg/d but bleeding remained controlled for the most part with intermittent fibrinogen replacement required for arterial line ooze. The patient was transferred back to the high dependency unit (HDU) on day 12. The patient had one readmission to ICU with diuretic-resistant fluid overload and hyponatraemia requiring intermittent haemodialysis. There were continued problems with clinically significant bleeding such as peripheral venous line oozing, the development of a chest wall haematoma and frank haematuria. Coagulation remained abnormal with a nadir PT of 36.9 seconds, APTT 109.9 seconds, fibrinogen 0.67 g/dL, platelets 35 × 103 μL. In total 3 pools of platelets, 10 g fibrinogen, 6 pools Octaplas® and 9 units red cells were administered over 48hrs. She was not considered as a suitable candidate for liver transplantation following multidisciplinary assessment. She was discharged to the HDU and died 5 days later.
Case 3
A 39 year old gentleman with decompensated cirrhosis and refractory ascites secondary to HCV awaiting OLT, was admitted to the ICU for CRRT for treatment of hyponatraemia resistant to medical therapy. He had a history of Haemophilia A and a preexisting coagulopathy in keeping with advanced liver disease but no clinically evident bleeding. Immediately prior to ICU admission the patient received 2 g of fibrinogen and 6000 IU Factor VIII and on admission, a further 6 g of fibrinogen were given to facilitate central venous access. Subsequently, CRRT was commenced without anticoagulant.
After initiating CRRT the patient became hypotensive necessitating vasopressor use. Clinically evident bleeding subsequently developed with a worsening coagulopathy requiring treatment as outlined in Table 3. Transfusion was aimed at maintaining a haemoglobin level above 8 gm/dL and other blood products given on an as required basis according to coagulation screen results and TEG performed during admission. Products transfused are detailed in Supplementary Table E3.
Despite aggressive replacement of blood products and other interventions to reduce blood loss including administration of tranexamic acid 1 gm 24 hours after admission, the patient continued to bleed profusely and remained severely coagulopathic. He also became profoundly hypotensive, requiring increasing doses of vasoactive medication. A worsening lactic acidosis developed along with resistant hyperkalaemia despite treatment with CRRT and repeated boluses of insulin and dextrose. The patient died 46hrs after admission despite maximal therapies.
Discussion
The above cases are as far as we can determine, the first reports of consumptive coagulopathy or disseminated intravascular coagulation (DIC) associated with initiation of CRRT in the ICU cohort of patients with decompensated cirrhosis. All three cases presented consecutively over a four month period to our unit and were placed on CRRT using the Prismaflex® AN69 ST150 membrane which had been recently introduced to our hospital. There were 19 additional patients with cirrhosis commenced on CRRT with the same membrane during this time period, without significant coagulation abnormalities. They highlight the precarious nature of the coagulation cascade in patients with advanced liver disease and potentially how easily this delicate balance can be upset. Furthermore, while the subsequent coagulopathy may resolve, it is also clear from these cases the potential for increased morbidity and mortality in this patient population.
The development of DIC in each case above was associated with a rapid derangement in coagulation parameters following initiation of CRRT with bleeding dominating the clinical picture. DIC is an acquired syndrome, occurring in up to 1% of hospitalized patients, and is secondary to an underlying disorder [14]. The mortality in ICU patients who develop DIC approaches 45% and is correlated with the severity of DIC and organ failure [15].
Decompensated cirrhosis in itself is not a condition associated with causing DIC. However, laboratory abnormalities can mimic DIC, with an elevated PT, INR, APTT and fibrin degradation products all of which were seen in the baseline coagulation screens from our patients. A relatively high factor VIII level and stable platelet count can be used distinguish the two [16]. In the cases described, falling platelet counts and low Factor VIII levels measured in Case 2 helped establish the diagnosis of DIC though this was more complicated to decipher in the Case 3 given the underlying Haemophilia A. The persistently dropping platelet and fibrinogen levels despite replacement in Case 1 helped distinguish between the coagulopathy associated with decompensated cirrhosis and DIC. Fibrinolysis is also thought to occur in cirrhosis with lower fibrinogen levels correlating with disease severity [6,16]. This is thought to be due to an elevated tissue plasminogen activator (tPA) level with a smaller increase in plasminogen activator inhibitor. However, when actual clot lysis is measured, for example using TEG, hyperfibrinolysis is either not evident or mild in otherwise stable patients [6]. In each case above fibrinogen, when measured, was above 1.0 g/L at baseline. To encapsulate both these phenomena and distinguish them from DIC, the term ‘accelerated intravascular coagulation and fibrinolysis’ (AICF) has been introduced [16]. It can be distinguished from DIC by the absence of frank bleeding and end organ damage [16]. When this normally balanced state is disrupted, DIC can develop. This balance is easily upset by factors such as infection (due to the production of endogenous heparinoids), renal failure, endothelial dysfunction and portal hypertension leading to clinically evident bleeding [4]. In Case 1, the patient was being treated for cellulitis at the time of ICU admission and the patient in Case 2 became pyrexial during the course of her ICU admission though this was considered related to blood transfusion. In Case 3, the patient was receiving antibiotics for a venous line infection. Theoretically, infection and production of endogenous heparinoids in some of these cases could have resulted in DIC but the temporal relationship of CRRT commencement and DIC potentially suggests an alternative or additive explanation.
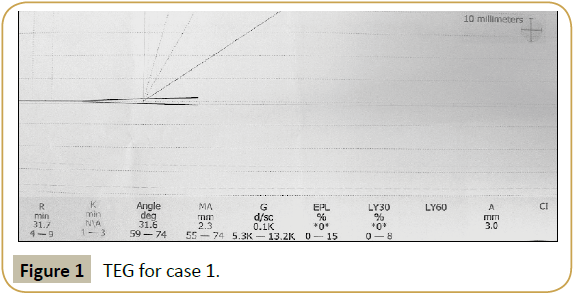
Figure 1:TEG for case 1.
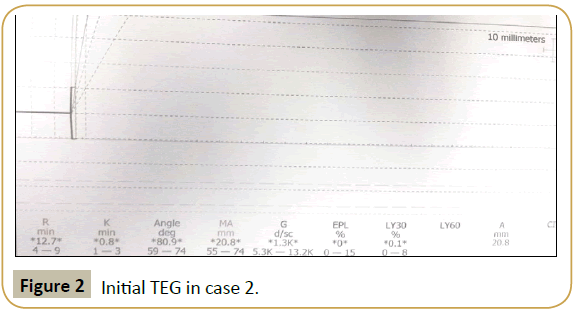
Figure 2:Initial TEG in case 2.
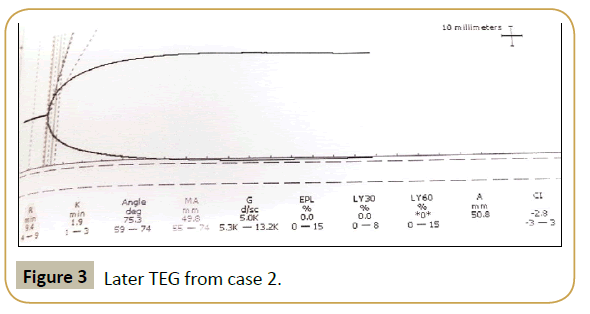
Figure 3:Later TEG from case 2.
| Time |
0 Hour |
6 Hours |
12 Hours |
24 Hours |
| Clinically evident bleeding |
nil |
Haematemesis |
Ooze from right groin vascath site
Bleed from nose and mouth |
Blood around cuff of Endotracheal tube
Ooze from mouth |
| Laboratory results |
Hb 9.1g/dL
PT 27.4
APTT 77.7
Platelets 82×103µL
Fibrinogen 1.11g/L
Factor VIII 22
Na+121mmol/L |
Hb 7g/dL
PT 32.7
APTT 171.8
Platelets 57×103mL
Fibrinogen 1.04g/L
Na+123mmol/L |
Hb 7.4g/dL
PT 33.2
APTT<240
Platelets 100×103µL
Fibrinogen 0.75gm/dL
Na+127mmol/L |
Hb 8.3g/dL
PT 26.6
APTT 240
Platelets 36×103µL
Fibrinogen 0.51g/L
Factor VIII 57
Na+131mmol/L |
| Treatment |
6gm fibrinogen |
2gm fibrinogen
2 pools platelets
2 units red cells |
6 units red cells
Fibrinogen 8gm
Octaplas® 12 pools
1 pool platelets
1000iu factor VIII and infusion 7000iu/24hrs
Factor VIIa 1000mcg
vitamin k 10mg
pressure to groin
nose packs
Tranexamic gauze to mouth |
3 units red cells
Fibrinogen 12gm
Octaplas® 8 pools
4000iu Factor VIII
2 pools platelets
Tranexamic acid 1gmadrenaline soaked gauze to mouth |
Table 3: Clinically evident bleeding compared to laboratory results and the treatments administered over time in hours from admission case 3.
A further feature separating these cases from the usual balanced state of haemostasis evident with cirrhosis was the marked hyperfibrinolysis present in each case and the persistently declining fibrinogen levels despite aggressive replacement. Hypofibrinogenaemia is only seen in very severe cases of DIC and fibrinogen can be normal in up to 57% of episodes [17].
DIC is a complex, systemic disorder that is characterized by widespread activation of coagulation, with fibrin deposition within vessels, consumption of clotting factors and end-organ damage and can manifest as either bleeding or thrombosis. A different subtype of DIC exists, most commonly encountered in patients diagnosed with Acute Promyelocytic Leukaemia (APML). In this subtype, hyperfibrinolysis predominates and coagulation factors, when measured, show a mainly haemostatic picture [18]. Significant hypofibrinogenaemia with haemorrhage dominate the clinical picture, while thrombotic complications are less frequent [18]. This primary hyperfibrinolysis is mainly the result of increased annexin A2 generation. Annexin A2 is a calcium dependant phospholipid binding protein, which increases tPA dependent plasmin generation 60-fold. In addition, there is increased degradation of fibrinogen by elastase, as also seen in sepsis [18]. The cases described above appear to share features of this DIC subtype, with haemostatic coagulation factors being documented in Case 2 and considerably reduced fibrinogen levels with problematic bleeding encountered in all three cases. We speculate that the deranged PT and APTT reflected hypofibrinogenaemia. We hypothesise that early fibrinogen replacement and inhibition of fibrinolysis in similar cases may be warranted.
Tranexamic acid, an antifibrinolytic agent which reversibly blocks the lysine binding sites on plasminogen, is not usually recommended as part of the treatment pathway for DIC [17]. The response to tranexamic acid in these cases was very pronounced with normalization of the TEG in Case 2 (Figure 2 and 3) and a reduction in clinically evident bleeding despite ongoing disturbance of the conventional coagulation profile. Antifibrinolytics have been used in the treatment of the DIC with hyperfibrinloysis subtype seen in APML [18]. Tranexamic acid has also been shown to attenuate the inflammatory response and reduce bleeding and transfusion requirements after cardiac surgery when given pre and post cardiopulmonary bypass (CPB) [19]. Given that the coagulopathy in the above cases was initiated by contact with an artificial membrane, it could be hypothesized that the treatment effect seen after CPB could be extrapolated to similar situations. However, the CRRT membrane has a much smaller surface area than the CPB circuit and the post pump or Kirklin syndrome [20], seen in around 10% of patients following CPB [21], occurs much less frequently in patients on CRRT. Extending the use of tranexamic acid outside its current therapeutic uses should only be recommended after rigorous clinical trials have been carried out to establish its safety and efficacy in this scenario.
The TEG and thromboelastometry measure viscoelastic changes in blood to give a global view of clot formation and dissolution. They more closely reflect in vivo haemostasis than traditional laboratory coagulation parameters and are useful as a point-ofcare testing device [22]. Thromboelastometry is more useful to detect thrombocytopenia and hypofibrinogenaemia and guide transfusion of platelets, fibrinogen and cryoprecipitate [23]. The use of thromboelastometry has been found to be useful to assess coagulation in patients with stable cirrhosis [24]. Risk of bleeding can be assessed by looking at the maximum clot firmness (MCF) and clot-formation time (CFT) which have good correlation with platelet number, antithrombin, factor II and fibrinogen levels [24]. MCF was also found to have a similar correlation to PT with Child- Pugh score in cirrhotic patients when determining prognosis [24]. TEG has also been shown to be of use in assessing bleeding risk and transfusion requirement in cirrhotic patients when clot strength in dynes/m2 is combined with INR and platelet count [25]. It can also be used to detect the presence of endogenous heparinoids which may contribute to the coagulopathy and haemorrhagic events associated with cirrhosis [26]. TEG has been shown to predict rebleeding in patients with variceal bleeding one day before haemorrhage recurred [27]. Parameters measured with TEG include the r and k times, α angle and maximum amplitude (MA). The r and k time measure the latency from the start of the test to initial fibrin formation and the time taken for the clot to reach amplitude of 20 mm respectively. They are representative of the traditional coagulation tests and prolongation reflects deficiency of clotting factors. The α angle measures the speed of clot strengthening while the MA represents the greatest strength of the clot and equates to platelet function and fibrinogen levels. LY30 and LY60 are additional parameters measured which give an indication of the degree of fibrin breakdown at 30 and 60 minutes after MA has been reached. An elevation of these indicates that hyperfibrinolysis is present. [28]. TEG was used in the above cases when automated laboratory coagulation screens were insufficiently precise to detect hyperfibrinolysis and guide treatment. The TEG pattern observed in Case 1 was similar to that described in patients rebleeding after variceal haemorrhage [27]. TEGs from Cases 2 and 3 had prolonged r times without the other features, suggesting the possibility that abnormalities of any of the three parameters could point towards increased risk of bleeding in patients with decompensated cirrhosis. Therefore it may be of benefit to perform a TEG in this patient population prior to interventions that carry the risk of inducing a worsening coagulopathy and/or bleeding. The TEG from Case 3 (Figure 4) was more difficult to interpret given the background of haemophilia.
Despite the classical picture of hyperfibrinolysis not being evident on the TEG from Case 2 (Figure 2), normalisation of TEG parameters after administration of tranexamic acid (Figure 3) may suggest it was actually present. This, combined with the haemostatic picture documented on clotting factor analysis suggests hyperfibrinolysis was the most likely cause of DIC. It has previously been suggested that TEG could be used to guide antifibrinolytic treatment in APML patients with hyperfibrinolysis, with therapy ceased once parameters normalize [18]. This could be another indication for TEG use in this patient type.
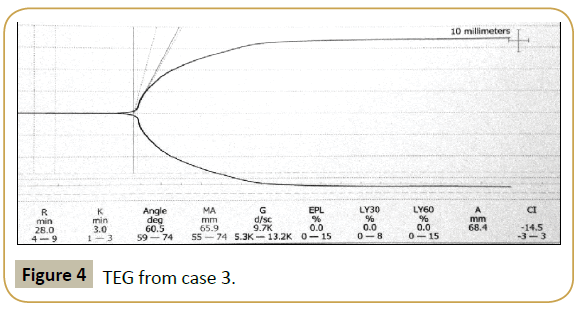
Figure 4:TEG from case 3.
In the three cases above we speculate that the initiation of CRRT in patients with a pre-existing coagulopathy secondary to decompensated cirrhosis, disrupted the delicate haemostatic balance of advanced liver disease resulting in DIC developing within 12 hours of starting CRRT.
It has been long understood that dialysis membranes have the potential to induce harmful effects when in contact with blood. This feature is referred to as biocompatibility. Their use induces contact phase activation and the subsequent reactions of coagulation. This usually results in blood clotting and this disruption of CRRT typically necessitates anticoagulation of the patient [29]. Due to the risk of bleeding and pre-existing coagulopathy in these cases no regional or systemic anticoagulant was administered. Artificial membranes can also activate polymorphonuclear cells, resulting in production of oxidative free radicals, which leads to inflammatory stress, endothelial cell activation and vascular injury [29], again resulting in activation of the coagulation cascade. It may have been either of these phenomena, initiating the reactions of coagulation, which resulted in bleeding rather than thrombosis in patients with an already deranged coagulation system.
Shortly prior to these three cases, the membranes used for CRRT were changed from the Prismaflex® HF1400 set to the Prismaflex® AN69 ST 150 set. Both membranes are synthetic, highly permeable, can bind endotoxins and cytokines and have improved biocompatibility over the older cellulose membranes [29]. Biocompatibility is characterized by both the zeta potential or electronegative charge of a membrane, a less negative charge being more biocompatible, and the amount of bradykinin generation. Production of bradykinin is a reflection of the degree of high molecular kininogen (HMWK) adsorption to the dialysis membrane and subsequent contact phase activation of the coagulation cascade therefore higher bradykinin levels implies reduced biocompatibility. The zeta potential of the AN69 ST150 is less than the HF1400 because it possesses a polyethyleneimine (PEI) coat. However, bradykinin generation is greater with the AN69 ST150 (Supplementary Table E4). Furthermore, in patients with decompensated cirrhosis, infection or bleeding and the production of endogenous heparinoids increases and may lead to DIC [4]. It is possible that these heparinoids could bind the AN69 ST150 membrane because of the positive charge on the PEI coat and later be released resulting in bleeding [30]. Perhaps the higher rate of HMWK adsorption and contact phase activation resulted in the development of DIC in combination with possible sepsis.
Conclusion
We report a consumptive coagulopathy developing in three consecutive patients following the initiation of CRRT in the setting of decompensated cirrhosis. Given the temporal relationship between initiation of CRRT and development of coagulopathy, there is a possibility that contact phase activation secondary to the CRRT membrane was a contributory factor in this process. It is likely, however, that the cause of DIC was multifactorial in these patients, with an element of sepsis and progression of underlying disease also playing a role. Whatever the exact mechanism of DIC in these patients, these cases emphasize that initiating CRRT in patients with decompensated cirrhosis requires careful consideration of the potential complications. This can include a rampant consumptive coagulopathy, necessitating aggressive replacement of blood products to treat and prevent catastrophic haemorrhage with significant morbidity and mortality associated.
References
- Blachier M, Leleu H, Peck-Radosavljevic M, Valla D-C, Roudot-Thoraval F (2013) The burden of liver disease in Europe: a review of available epidemiological data. J Hepatol 58: 593-608.
- Lim YS, WR Kim (2008)The global impact of hepatic fibrosis and end-stage liver disease. Clin Liver Dis 12: 733-746.
- JG F (2002) Hepatic encephalopathy, hepatopulmonary syndromes, hepatorenal syndrome, coagulopathy and endocrine complications of liver disease. In: Feldman M, Friedman LS, Sleisenger MH (edr)Sleisenger&Fordtran’s Gastrointestinal and Liver Disease: Pathophysiology/Diagnosis/Management. Pa Saunders, Philadelphia.
- Tripodi A, PM Mannucci (2011)The coagulopathy of chronic liver disease. N Engl J Med 365: 147-156.
- Northup PG, Caldwell SH (2013) Coagulation in Liver Disease: A Guide for the Clinician. ClinGastroenterolHepatol 11: 1064-1074.
- M Senzolo, P Burra, E Cholongitas, AK Burroughs (2006) New insights into the coagulopathy of liver disease and liver transplantation. World J Gastroenterol 12: 7725-7736.
- Ciccone MM, Principi M, Ierardi E, Di Leo A, Ricci G, et al. (2015) Inflammatory bowel disease, liver diseases and endothelial function: is there a linkage? Review J Cardiovasc Med (Hagerstown) 16:11-21.
- Michele B, Maria TV, Annabianca A, Serafina S, Annapaola Z, et al. (2015) Endothelial Dysfunction Correlates with Liver Fibrosis in Chronic HCV Infection. Gastroenterol Res Pract 2015: 682174.
- Biggins SW, Rodriguez HJ, Bacchetti P, Bass NM, Roberts JP, et al. (2005) Serum sodium predicts mortality in patients listed for liver transplantation. Hepatology 41: 32-39.
- Arroyo V, Ginès P, Gerbes AL, Dudley FJ, Gentilini P, et al. (1996) Definition and diagnostic criteria of refractory ascites and hepatorenal syndrome in cirrhosis. International Ascites Club,Hepatology 23: 164-176.
- Asrani SK, WR Kim (2010) Organ allocation for chronic liver disease: model for end-stage liver disease and beyond. CurrOpinGastroenterol 26: 209-213.
- Dawwas MF, Lewsey JD, Neuberger JM, Gimson AE (2007) The impact of serum sodium concentration on mortality after liver transplantation: a cohort multicenter study. Liver Transpl13: 1115-1124.
- Yun BC, Kim WR, Benson JT, Biggins SW, Therneau TM, et al. (2009) Impact of pretransplanthyponatremia on outcome following liver transplantation. Hepatology 49: 1610-1615.
- Levi M, Cate HT (1999) Disseminated intravascular coagulation. N Engl J Med 341: 586-592.
- Okabayashi K, Wada H, Ohta S, Shiku H, Nobori T, et al. (2004) Hemostatic markers and the sepsis-related organ failure assessment score in patients with disseminated intravascular coagulation in an intensive care unit. Am J Hematol 76: 225-229.
- Caldwell SH, Hoffman M, Lisman T, Macik BG, Northup PG, et al. (2006) Coagulation disorders and hemostasis in liver disease: pathophysiology and critical assessment of current management. Hepatology 44: 1039-1046.
- Levi M, Toh CH, Thachil J, Watson HG (2009) Guidelines for the diagnosis and management of disseminated intravascular coagulation. Br J Haematol 145: 24-33.
- Breen KA, Grimwade D, Hunt BJ (2011)The pathogenesis and management of the coagulopathy of acute promyelocyticleukaemia. Br J Haematol 156: 24-36.
- Jimenez JJ, Iribarren JL, Lorente L, Rodriguez JM, Hernandez D, et al. (2007) Tranexamic acid attenuates inflammatory response in cardiopulmonary bypass surgery through blockade of fibrinolysis: a case control study followed by a randomized double-blind controlled trial. Critical Care 11: R117.
- Kirklin JK, Westaby S, Blackstone EH, Kirklin JW, Chenoweth DE, et al. (1983) Complement and the damaging effects of cardiopulmonary bypass. J ThoracCardiovascSurg 86:845-857.
- Cremer J, Martin M, Redl H, Bahrami S, Abraham C, et al. (1996) Systemic inflammatory response syndrome after cardiac operations. Ann ThoracSurg 61:1714-1720.
- Herbstreit F, Winter EM, Peters J, Hartmann M (2010) Monitoring of haemostasis in liver transplantation: comparison of laboratory based and point of care tests. Anaesthesia 65: 44-49.
- Roullet S, Pillot J, Freyburger G, Biais M, Quinart A, et al. (2010) Rotation thromboelastometry detects thrombocytopenia and hypofibrinogenaemia during orthotopic liver transplantation. Br J Anaesth 104: 422-428.
- Tripodi A, Primignani M, Chantarangkul V, Viscardi Y, Dell'Era A, et al. (2009) The coagulopathy of cirrhosis assessed by thromboelastometry and its correlation with conventional coagulation parameters. Thromb Res 124: 132-136.
- Shah NL, Xavier E, Northup PG, Patrick GN, Gavin S, et al. (2009)The use of thromboelastography, platelets and INR in a clinical model for bleeding risk in cirrhotic patients. Gastroenterology 136:A795-A796.
- Mucino-Bermejo J, Carrillo-Esper R, Uribe M, Mendez-Sanchez N (2013) Coagulation Abnormalities in the Cirrhotic patient. Ann Hepatol 12: 713-724.
- Chau TN, Chan YW, Patch D, Tokunaga S, Greenslade L, et al. (1998)Thrombelastographic changes and early rebleeding in cirrhotic patients with variceal bleeding. Gut 43: 267-271.
- Thakur M, Ahmed AB (2012) A review of thromboelastography. International Journal of perioperative ultrasound and applied technologies 1: 25-29.
- Chanard J, Lavaud S, Randoux C, Rieu P (2003) New insights in dialysis membrane biocompatibility: relevance of adsorption properties and heparin binding. Nephrol Dial Transplant 18: 252-257.
- Chanard J, Lavaud S, Maheut J, Kazes I, Vitry F, et al. (2003) The clinical evaluation of low-dose heparin in haemodialysis: a prospective study using the heparin-coated AN69 ST membrane. Nephrol Dial Transplant 23: 2003-2009.