Keywords
Barriers to cessation; Reasons for quitting; Cigarette; Drinking; Concurrent
Introduction
Embryonic stem cells (ESC’s) are a set of pluripotent cells unique in character which are obtained from preimplantation blastocyst stage embryos. They can either undergo asymmetric divisions whereby they either duplicate themselves or differentiate into another cell type. Adult stem cells on the other hand are undifferentiated cells found around differentiated cells in a tissue or an organ. While they are multipotent they can differentiate into a limited number of cell types. ESC’s can proliferate indefinitely in an undifferentiated state [1]. They express specific markers or characteristics like stage specific embryonic antigens, enzymatic activities like alkaline phosphatase and telomerase, ’stemness’ genes which are rapidly downregulated upon differentiation including Octamer (Oct 4) and Nano g. On the other hand they can differentiate in vivo in teratomas in cells representing the three major germ layers; endoderm, ectoderm, mesoderm or they can be directed into differentiation in vitro into any of the 200+cell types present in the adult body. Many human diseases result from defects in a single cell type, the potential to replace defective cells by cells or tissue replacement therapy involving differentiated human ESC (hESC) provides a possible cure or at least the alleviation of symptoms of various degenerative diseases or into the production of substantia nigral dopaminergic neuronal phenotype with potential application in the treatment of Parkinson’s disease. Other potential applications of stem cell therapy include strokes, Alzheimer’s disease, heart disease, osteoporosis, insulin dependent diabetes, leukemia, burns, spinal cord injury. Currently the ongoing safety and therapeutic efficacy of parthenogenetic derived stem cells has begun, with food and drug administration approving the use in macular degeneration in humans [2,3]. In this review we describe the characterisation of ESC, cloned ES cells –advantages and disadvantages of using ESC’S from conventional embryos, and role of parthengenetic ESC’S derived by artificial activation of oocytes left redundant due to failed fertilization or chromosomal abnormalities. This offers enormous advantages and obviates the ethical issues involved along with recent studies in primate where probably immunogenicity may have been overcome with no need of HLA typing. A brief report on germ cell development from spermatogonial stem cells to develop gametes in prepubescent boys where cryopreservation may not be possible before the exposure to gonadotoxic chemotherapy is also discussed. Further the recent advances in use of neural stem cells by induced pluripotent stem cells In Alzheimer’ disease is also discussed.
Derivation and Culture of ES cells
ES cells were first established from preimplantation murine embryos i.e. in 1942. They were derived from inner cell mass (ICM) of expanded blastocyst. For establishing ES cells, ICM is isolated by immunosurgery to remove trophoblasts cells. After several day in culture isolated ICM cells form a colony which can be expanded by disaggregating and reseeding on non-proliferative mitomycin C treated/irradiated fibroblasts(STO cells or primary mouse embryonic fibroblasts [4,5]. To prevent spontaneous differentiation, ES cells must be maintained by repeat passages on feeder layers, usually a feeder layer generally required to isolate ES cells and to support their successive passages [6]. Main role of feeder cells is probably to provide growth factors necessary for proliferation and inhibition of spontaneous differentiation. The principal differentiation inhibitory factor is leukemia inhibitory factor (LIF), as demonstrated by the fact that LIF-defective fibroblasts cannot maintain ES cells in undifferentiated state [7] and LIF in the medium can support ES cells without feeder cells [6,8]. LIF is a pleiotropic cytokine similar to ciliary neutrotrophic factor [9], oncostatin M [10], and other related cytokines acting through gp130 like interleukin 6 [11]. Standard culture conditions contain fetal bovine serum although it is not well characterized and is susceptible to batch-to-batch consistency.
Besides mouse models isolation of ES cells has been attempted on rats, mink, rabbits, hamster, primates, sheep, cattle and pigs. In 1998, human ESC were first isolated from in vitro fertilized blastocysts [12], using mouse embryonic feeder cells and serum containing medium. Human ESC is typically cultured with animal derived serum or serum replacement in mouse feeder cells. Culturing human ESC’s with serum replacement on mouse feeder cells are the sources of the nonhuman Salic acid Neu5Gc as demonstrated, which could induce an immune response upon transplantation of hESC into patients [13]. The use of feeder free systems such as Matrigel or other components of the extracellular matrices have been explored [14,15]. However, matrix components used for feeder free culture are still from animal source and the medium also contains animal derived products. More hESC lines are being established with the numbers growing abruptly [16]. Derivation of immune-compromised hESC cells using somatic cell nuclear transfer was considered a breakthrough in hESC cell research in 2004 [17].
Therapeutic Cloning
Cibelli et al. tried development of cloned human embryos to 8-10 cell stage but failed to develop blastocysts for derivation of human cloned ES cells [18]. Dr Hwang’s lab in 2004 derived the first successful human ESC from cloned blastocyst, after optimizing the nuclear transfer protocol to obtain a cloned human blastocyst with the development rate of 29% [17]. They started from scratch with no previous successful human parameters to work with, they allowed fused donor cell nucleus to reprogram on side of the enucleated oocyte for 2 hrs activated with 10 μM ionophore × 5’/DMAP cultured × 4 h then cloned embryo for 48 h in G1. 2, media and then transferred them into human modified synthetic oviductal fluid (SOF supplemented with amino acids-SOFaa) for the rest of their in vitro development [17]. Dr. Hwang besides these used very fresh oocytes, donated from fertile women, and barely allowed them to mature prior to enucleation, utilizing a very gentle squeezing technique, to cause less damage as compared to typical aspiration [17]. With this improved technique, he could obtain significant quantities of cloned blastocyst, deriving ntESC lines from, the overall efficiency of which was very low with 242 oocytes used to derive one ntESC line [17]. To improve ntESC derivation efficiency Dr. Hwang made sincere efforts by optimization of experimental protocol, which included using human feeder cells limiting trypsin exposure of donor cells to 30 seconds while monitoring for cellular damage and keeping the oocyte hylaluronidase to a minimum, and thus deriving ESC’s directly from the nuclear transfer blastocysts rather than using immune surgery and performing large amounts of practice nuclear transfer (typically using non-human oocytes to improve his technicians micromanipulations skills [19]. With this superior protocol Dr. Hwang managed to get on average 1 ntESC line/every 12 human oocytes used. He also showed that each ntESC line he derived was pluripotent, chromosomally normal and expressed the exact immunologically identical MHC antigens as the donor patients [19]. Although with these many advances the issues which need to be resolved are 1) Humphrey et al. showed that cloned mice show significant aberrant gene expression as compared to IVF ES cells, and if so by how much [20]? Human ntESC demonstrated same morphology, stem cell markers as IVF ESC [19]. 2nd ntESC possess the same nuclear gene as donor but express mitochondrial epitopes from enucleated oocyte’s mitochondrial DNA. That mitochondrial heteroplasmy could influence ntESC‘s ability and differentiation [21], and hence further research is required. Although Lanza et al. could not find any rejection of bovine differentiated ntESC following transplantation-even though they expressed allogenic mitochondrial alleles [22], which suggests that mitochondrial heteroplasmy does not have significant negative effect. Other factors to be investigated include the presence of lack of sperm induced centrosome, in ntESC’s, effect of non-random X inactivation in ntESC and the possibility of shortened telomeres of their somatic donor source [19].
Can Therapeutic Cloning be Used Medically?
Despite Dr. Hwang’s production of ntESC’s can help in treating a patient with diabetes type 1 as cloned ntESC’s would be producing same defective insulin gene, hence ntESC’s would have to undergo a round of gene therapy to correct the genetic defect in question. In this type 1 DM the ntESC would need an insertion (via homologous recombination of functioning insulin gene before being differentiated and transplanted back into the diabetic patient. Therapeutic cloning with gene therapy has been successfully performed in mouse model [23]. Rag2-/-mice are severely immunodeficient due to their inability to produce lymphocytes. Rideout et al.; produced murine ntESC from Rag2- /-immunodeficient mice and then replaced the defective Rag 2 allele in the ntESC with a functional Rag2 allele via homologous recombination. These genetically corrected ntESC s were then differentiated into haematopoietic precursors and transplanted back into Rag2-/-mice [23]. This gives evidence for treatment of certain genetic diseases using a combination of therapeutic cloning and gene therapy. Similarly, murine ntESC’s have also been used to cure murine equivalent of Parkinson’s diseases [24]. Barberi et al. differentiated murine ntESC-derived dopaminergic neurons into Parkinson’s mice. The mice showed a significant alleviation of their Parkinsonism phenotype with no aberrant differentiation and no teratocarcinoma formation was observed [24]. Thus, both Rideout and Barberis study demonstrate that therapeutic cloning along with gene therapy if needed can be used to cure diseases in murine models. This lays foundation for treatment of human regenerative diseases.
Role of Parthenogenetic Embryonic Stem Cells
Parthenogenesis is a reproduction strategy in which no sperm is involved to trigger embryonic development from the oocyte and the female generates an offspring without any paternal inheritance [25] with parthenogenesis coming from the Greek word virgin birth and examples of it being in whiptail lizard and virgin birth of shark in captivity [26]. Although in animals parthenogenesisis is not a natural form of reproduction, genetic modification technologies have made it possible for the creation and birth of fertile animals from mouse parthenogenetically activated oocytes [27]. Interest has developed in parthenogenetic activation of mammalian oocytes because 1) Experimental work that involves parthenogenetically developed embryos circumvents ethical and legal problems concerning use of human embryos which are generated for reproductive purposes. 2) These parthenotes may be involved for various ART related research studies, on human pluripotent stem cells or basic science, which command human embryonic development as well as cloning experiments using somatic cell nuclear transfer in mammalian oocytes. 3) The creation of graded parthenogenetic human ESC (hpESC) lines has the potential to benefit a vast number of patients, when used in cell therapies, with a reduced chance of transplant rejection. However this will only be possible if clinicians participate in such research projects and enough clinical grade hESC lines for use in cell/tissue therapies are there, if research laboratories obtain enough biological material from ART centres to evaluate activation strategies, derivation, culture media are adequate for future therapeutic use of phESC lines.
Techniques Used for Pathenogenetic Activation
Oocyte activation
Non-fertilized metaphase II oocyte will remain arrested in this stage till a stimulus, either in the form of a fertilizing spermatozoan or some artificial agent triggers increase in intracytoplasmic Ca2+ rises and initiates meiosis resumption. These intracellular Ca2+oscillations inhibit the action of metaphase promoting factor (MPF) as well as the cytostatic factor (CSF) and lead to metaphase/ anaphase transition, segregation of sister chromatids, along with extrusion of the second polar body. On preventing the elevation of intracellular Ca2+ after sperm penetration by preloading the oocytes with Ca2+ chelator BAPTA1AM [28]. In the absence of intracellular Ca2+ increase, and activation, embryo development failed to occur highlighting the importance of these calcium transients. In mammals it is the specific phospholipace C zeta (plc-zeta) or the post acrosomal sheath WW domain binding protein(PAWP)released by the sperm during the normal fertilization process [29]. Activating agents used may mimic the sperm bound stimuli to release Ca from the endoplasmic reticulum and exit from meiotic arrest and start embryonic development.
Agents used
Except for strontium (Sr2+) [27], all other artificial agents studied do not produce repetitive Ca2+ oscillations, normally observed during fertilization. Although the single rise in intracellular Ca2+, produced by agents like ionomycin or ethanol is adequate for triggering meiotic resumption along with cortical granule release. Specific oocyte activation protocols using strontium chloride (SrCl2) to induce continuous Ca2+ oscillations during exit from 2nd meiotic and first embryonic mitosis events seem to have a role in long term embryonic events, such as number of cells in the inner cell mass (ICM )and trophectoderm of the resulting blastocyst [29].
Three possible parthenenotes may result
• Treatment with an activating agent like SrCl2, ethanol, Ca2+ionophore or ionomycin is followed by a protein synthesis inhibitor like dimethylaminopurine (DMAP-a broad protein synthesis inhibitor) or cytochalasin B or D (inhibitors of actin filaments polymerization) to block the second polar body extrusion. Hence, the parthenote will be a pseudodiploid heterozygote embryo, which contains two sister chromatids of each maternal chromosome present in the MII oocyte.
• Second methodology may allow extrusion of second polar body, with the existing parthenote containing a single copy of the sister chromatids, which undergoes spontaneous diploidization and hence resulting in a completely homozygous diploid parthenote.
• Activating agent induces exit from MII and without second polar body extrusion, without diploidization, leading to a haploid zygote, developing into a haploid parthenote. Unfortunately, in this instance, embryo development into blastocyst stage is very low.
For getting mouse oocytes activation, oocytes are exposed to Ca2+ free medium supplemented with 1 mM SrCl2X2h [27,30]. The same doesn’t work well with bovine oocytes, where high activation rates achieved by exposing oocytes to ionomycin x 5 min followed by 2 mM 6-dimethylaminourine (DAMP) x 3 5h [31]. This method has been used to get high rates of bovine pESC colonies [32]. It is essential to give three broad protein synthesis or kinase inhibitors like 6 DMAP, cycloheximide or roscovitine [33], to maintain inactivation of meiotic kinases as although initial decline in meiotic kinase activities occurs with Ca ionophores like ionomycin MAP can soon recover leading to the oocyte entering into meiotic arrest, which is known as metaphase III [34].
Activation in Human Oocytes
Aim has been to generate blastocysts for creation of phESC lines [35]. These parthenogenetic embryos and ESC deficiencies parallel to those of sperm fertilized counterparts [36]. Right after first phESC lines, these lines have been created using Ca2+ ionomycin along with 6 DMAP [37], although Mai et al. combined electrical and chemical stimuli using ionomycin and 6 DMAP for the artificial activation of the oocytes. Although their parthenogenetic blastocyst rate was lower than the first one in which 23 blastocysts, 50% were generated from 6 phESC lines, i.e. they got only 21%, the ICM of 3 blastocysts got used for successful derivation of new parthenogenetic stem cell lines. Then one has to take into account that normally also in IVF /ART cycles blastocyst formation Rates vary with patient to patient protocols with patients age, sperm parameters, culture media used, ovarian stimulation protocols, hence same is to be expected regarding development of a Parthenogenetic blastocysts with different protocols and donors of oocytes or oocyte conditions.
During an attempt to develop spindle transfer (ST) oocytes for mitochondrial DNA replacement and develop ESC’s Tachibana et al. found that unfertilized metaphase II oocytes from mammals are sensitive to mechanical/physical stimuli where pressure, osmolarity, flux or temperature changes can cause spontaneous activation in meiotic resumption and parthenogenetic development [38,39].
Activation of Failure to Fertilize (FF) Human Oocytes
Although recue ICSI has been commonly advocated within few hours of detection as a treatment following failed fertilized oocytes [40-43], risk of chromosomal abnormalities remains in the resulting embryo with the oocyte ageing since the collection time increases, markedly by the time ICSI is undertaken, sometimes it may be as high as over 30hour or >after OPU, with varying gestations attributed by various centres, although not carried out by all centres.
However the Sneddon et al. studied the developmental potential of these FF oocytes and developed 8 blastocysts from 579 clinically failed eggs-these 8 blastocysts originated from 2 abnormally fertilized and 6 FF, with 3 pronuclei stage (abnormally fertilized, which were parthenogenetically activated by exposure to ionomycin and followed by 6DMAP along with a protein synthesis inhibitor cycloheximide. The gene expression profiling and developmental potential was similar as the normally fertilized ones [44,45] (Figure 1). This makes these unwanted artificially activated oocytes an alternative source for the generation of human ESC’s lines.
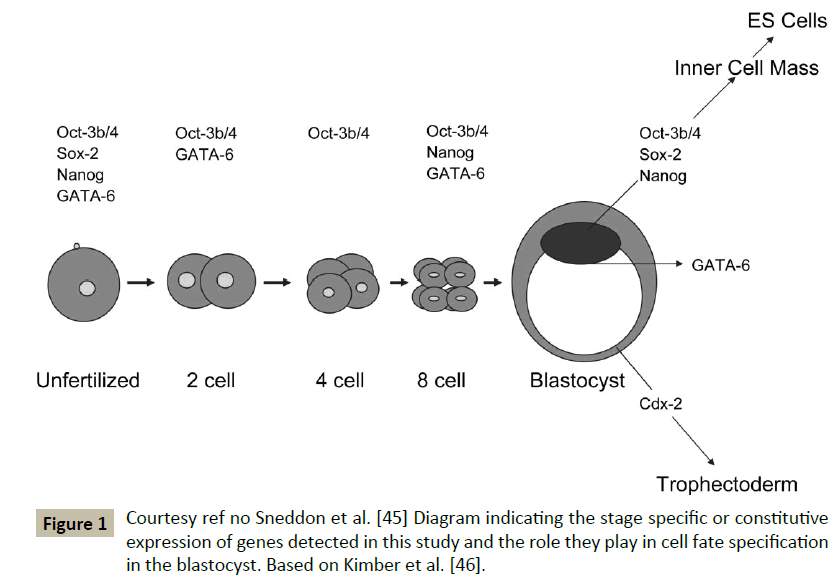
Figure 1: Courtesy ref no Sneddon et al. [45] Diagram indicating the stage specific or constitutive expression of genes detected in this study and the role they play in cell fate specification in the blastocyst. Based on Kimber et al. [46].
Kyono et al. obtained very high fertilization rates by using SrCl2 in patients having previous very low fertilization rates. They exposed oocytes to SrCl2 immediately after insemination by ICSI and fertilization rates increased from 27% in previous cycles to 64%, with 6 pregnancies ensuing, of which 4 went to term. With 5 live children with this procedure no neurological/physical deficit was found [46]. Similarly, he used a globozoospermic sperm and a Ca2+ ionophore to generate zygotes [47]. Other groups have replicated these findings for FF [48-52]. Still more studies that are epigenetic are warranted, with long term follow up of children.
Epigenetics Imprinting and Parthenogenesis
The reason why mammalian parthenogenesis is not supported in contrast to other female vertebrates is that the mammalian specific genome imprinting, is a deviation from Mendelian inheritance where parents of origin leave epigenetic marks which results in strictly paternal or maternal expression of genes [53,54]. Parthenotes lack sperm alleles, hence paternally expressed genes are functionally absent. On the otherhand maternally expressed imprinted genes are transcribed from both alleles which leads to overexpression and is associated with developmental abnormalities. Sritanaudomchai et al. further showed the down regulation of a novel paternally expressed imprinted gene inosoitol phosphate phosphatase(INPPSC) in PESC besides 12 highly downregulated putative paternal genes [55]. Many imprinted genes are directly involved in fetal growth pathway, the wide genome dosage imbalance serves as a barrier for normal fetal development in parthenotes [53]. Surani et al. studied hat monoallelic gene expression from paternal genome is critical for extraembryonic tissue formation and function [56]. Like paternally expressed Igf2 and its maternal regulators Igfr2, Grb10 and H19 are critical for placental growth and nutrient diffusion [57-63].
Biparental reproduction is necessary because of parent specific epigenetic modification of the genome during gametogenesis. Unusual expression of imprinted genes from maternal and paternal allelles results due to this. Kono et al. used reconstructed oocytes, both mature as well immature containing 2 haploid sets of maternal genome by appropriate expression of igf2 and h19 genes along with other imprinted genes using mutant mice with a 13 kilobase deletion in h19 gene as non-growing donor. This parthenote developed to adulthood with ability to reproduce suggesting paternal imprinting prevents parthenogenesis and ensuring that Paternal contribution is obligatory for the descendant [27].
Viable bilateral mice the first sperm free mice were generated by Kono et al. by combining genome of 2 unrelated oocytes, surviving 293 days. An on-growing oocyte from a newborn mouse having undergone complete imprint erasure without developing established maternal imprint signatures [27]. Kawahara et al. to mimic paternal silencing genes, the paternal igf2-H19 and Dlk1-Gtl2on chromosome 7 and 12-2 differentially methylated regions deleted them in imprint free oocytes. This was followed by nuclear transfer and fusing with mature imprinted oocytes which allowed them to reconstruct nuclei from these imprint free oocytes and then activate them subsequently artificially allowing bimanual parthenogenetic embryos developing into a live offspring [64]. Thus correcting few imprinted domains could rescue parthenogenetic development. This was the reason given for the longevity of female is species because of regulating longevity and energy conserving genes [65]. The offspring’s produced thus were 30% lighter in weight but had a longer life span [65]. Still though removal of 2 paternally imprinted regions allowed normal development in mouse models but their maybe other epigenetic aberrations in mammalian parthenote. In humans e.g. children with uniparental imprinting developmental disorders like Silver russel, Beckman-Wiedman, Angleman syndrome display acute developmental defects varying from mental retardation to physical abnormalities [66].
Trying to determine if genomic imprinting can be maintained during process of ES cell derivation from pig blastocysts Uh et al. analysed in 1 IVF and 3 PG established cells and found expression of H19 gene was significantly greater In PG blastocyst as compared to IVF blastocyst but IGF2 was greater in IVF than PG blastocyst. Although mRNA varied in PG cell lines the IGF2-H19 had a differentially methylated region (DMR)3, which was typically unmethylated in all PG cells and hemimethylated in IVF cells suggesting DNA methylation status is constant although mRNA of H-19 and IGF2 gene is susceptible in vitro environments during the process of ES cell derivation from blastocyst [67].
Potential Therapeutic Uses for Regenerative Medicine
The uniparental origin of parthenogenetic SC was utilized to develop PESC toward cardiac lineage and applied to tissue engineered cardiac repair and proposed subsequently for human myocardial tissue engineering in cases of myocardial infarction after successful murine experiments using development of PSC and then transformation into cardiomyocytes and in view of acceptance of PSC in MHC matched allotransplantation. Further they proposed once human PSC are widely available they presumed these PSC’s would be amenable to myocardial tissue engineering just as hESC or iPSC are [68,69] as proven by their murine experiments where they enriched cardiomyocytes, to facilitate engineering of force generating myocardium and enhancing regional myocardial function in MI [70].
Although human ESC are derived from spare IV4F embryos, they would be genetically divergent from any patient requiring tissue transplantation and are likely to incite rejection unless immunogenic drugs are used. Hence, till now current use is limited to immune privileged sites like CNS or the eye. Nuclear transfer, which intends to produce ESC’s genetically identical to the patient by SCNT, may avoid immune rejection, which has been recently achieved in primates [71]. But a very high cost along with low efficiency makes one look for alternative sources of histocompatible pluripotent cells. Diploid Monkey Parthenote embryos have been generated by artificial activation of metaphase II arrested oocytes followed by retention of 2nd polar body [34]. Although such embryos offer a potential source of pluripotent cells and would be isogenic with the donor [72,73], the potential of these fetuses to implant /complete development is debatable. To form viable fetuses [74,56]. Although this overcomes the ethical issue regarding derivation of human ESC ‘s Because of Destruction of Potential life, some concerns remain regarding aberrant genomic printing, high levels of homozygosity. For e.g. disruption if imprinted genes or their inappropriate expression is associated with certain severe syndromes/carcinogenesis in humans [75]. Dighe et al. demonstrated in 5 rhesus monkeys that PESC are genetically and epigenetically variable and carry some lines which attenuate many of these concerns and lot of banks can be considered to be practical and suitable for autologous transplantation being cost effective [76]. Although PESC are morphologically indistinguishable from biparental controls the parthenogenetic embryos maintain aberrant imprints and thus cannot develop to term but established PEC’s can correct some of these defects and display normal gene expression [76,77]. One important characteristic of phESC is the fact that they show frequent homozygosities in the major histocompatibility locus, which may allow efficient immune matching [77].
For regenerative medicine, potential of human PESC’s is evident by studies, which demonstrated that similar in vivo and in vitro differential potential of ESC’s exists. Both human and nonhuman primate PESC readily form teratomas, when injected into immunodeficient mice, which are indistinguishable from composition from biparental controls [37,55,73,76,78].
Espejel et al. demonstrated that PESC differentiated into hepatocytes providing normal liver function in viable adult mice in case with lethal liver failure due to fumaryl acetoacetate hydrolase (Fas) [79]. In mice, PESC derivatives supported longterm haematopoiesis as well [80]. While in primate models PESC have been differentiated into cardiomyocytes as mentioned earlier [70], as well as dopamine neurons which caused long term survival once transplanted in to brain allografts without any teratoma formation [81].
Therapeutic Actions of iPS
Generating disease specific and patient specific iPS cells through reprogramming has become routine. Mechanistic insights into a variety of diseases to carry out in vitro drug screening to evaluate potential therapeutic and to explore gene repair
• Lee et al. used iPS cells to demonstrate disease modelling and drug screening for familial dyautonomia, caused by a single point mutation in the gene encoding the inhibitor of NF?B (I?B)-kinase complex associated protein (IKBKAP) which manifest as an extensive defect of ANS and dysfunction in small fibre sensory neurons. With the development iPS cells from patients with familial dysautonomia, investigators produced peripheral and central nervous system precursors and found three disease related phenotypes. After screening with multiple compounds, they showed that the kinetin could partially normalize the abnormal phenotype, which is a plant hormone [82].
• Several research groups have generated models of long QT syndrome, a congenital disease with 12 types, each of which is associated with abnormal ion channel function, a prolonged QT interval on an ECG and a high risk of sudden cardiac death due to ventricular fibrillation. In animal models lot of work has been done to study the underlying mechanisms of this syndrome, but cardiomyocytes having distinct electrophysiological properties which differ between species, lack of in vitro sources of human cardiomyocytes along with difficulty in modeling patient specific variations impeded studying this.
• Moretti et al. differentiated iPS cells from individuals with type 1 long QT syndrome into cardiomyocytes and derived long prolonged action potentials in the ventricular and atrial cells as predicted [83]. With the use of this model they uncovered a dominant negative trafficking defect associated with particular mutation that causes this variant of long QT syndrome. Further, these cardiomyocytes had increased susceptibility to catecholamine induced arrhythmias, and compounds which exacerbated the condition e. g. isoprenaline were identified. Hence treatment of these cardiomyocytes withβ-adrenergic receptor blockers attenuated the long QT phenotype.
• Itzhaki et al. modelled type 2 long QT Syndrome in cardiomyocyes. They found that the long QT syndrome phenotype was aggravated by blockers of ERG-type potassium channels, whereas nifedipine, a calcium channel blocker and pinaclidil, an agonist of the ATP sensitive potassium channel, both ameliorated the long QT syndrome phenotype, as shown by decreased duration of action potentials in long QT syndrome cardiomyocytes, and eliminated early after depolarizations and the abolishment of all triggered arrhythmias. Limitation of this is excessive shortening of the action potential duration, leading to short QT syndrome [84].
• Aggarwal et al. explored a condition of telomere maintenance, known as dyskeratosis congenital, where they found, in its most severe form, dyskeratosis congenital is caused by a mutation in the dyskerin gene (DKC1), is X-linked, leads to shortened telomeres and premature senescence in cells and ultimately manifesting as degeneration of multiple tissues. The induction of iPS cells by reprogramming is accompanied by the induction of gene telomere reverse transcriptase (TERT), it did not limit the development and maintenance of iPS cells from individuals with dyskeratosis congenital. Despite the efficacy of iPS cells being poor, the authors could successfully reprogram patient fibroblast. Although the telomere length was immediately after reprogramming was shorter than that of the parental fibroblast population, continuous passage of some iPS cell lines has led to telomere elongation over time. This was accompanied by upregulation of expression of TERC, which encodes the RNA subunit of telomerase [85].
Further it was shown that TERT, TERC as well as DKC1,2 were expressed higher at higher levels in dyskeratosis-congenitaderived iPS cells than in parental fibroblasts. The authors determined that the genes encoding the components of the telomerase pathway, including a cis element of the 3 regions of the TERC locus, which is essential for a transcriptionally active chromatin structure-were direct binding targets of the pluripotency associated transcription factors. On further analysis, they found that the transcriptional silencing owing to a 3’ deletion in the TERC locus leads to autosomal dominant form of dyskeratosis congenital by diminishing TERC transcription. Though telomere length is restored in dyskeratosis-congenita-derived iPS cells, differentiation into somatic cells is accompanied by a return to pathogenesis with low TERC expression and decay in telomere length. This shows that TERC RNA levels are dynamically regulated and that the pluripotent state of the cells is reversible, suggesting that drugs which elevate or stabilize TERC expression might rescue defective telomerase activity and provide therapeutic benefit [85].
• In an independent study of reprogramming of cells from patients with dyskeratosis congenital Batista et al. [86], confirmed the general transcriptional upregulation of multiple telomerase components and the maintenance of telomere lengths in many clones [86], however In this study no clones with elongated telomeres were identified. The different outcomes in 2 studies show the limitations of iPS cell based disease models which are imposed by clonal variation, due to technical infidelity of reprogramming [87]. Further before a given iPS cell model can be claimed truly representative of the disease, how many patients must be involved, and how many iPS cell lines must be derived from each patient. Hence, all these issues need to be kept in mind while generating disease models and making claims on results from these models.
• Besides using iPS cells for modeling diseases in vitro, the goal of developing patient specific stem cells has been motivated by the prospect of generating a ready supply of immune compatible cells and tissues for autologous transplantation. At present this clinical translation of iPS-cell based therapies seems more futuristic than the in vitro use of iPS cells for drug development but two studies have provided the proof in mouse models that the dream might one day be realized by Jaerisch et al. who used homologous recombination to repair the genetic defect in iPS cells derived from a humanized model of sickle cell anaemia [88]. Directed differentiation of the repaired iPS cells into haematopoietic progenitors followed by transplantation of these cells into the affected mice led to the rescue of the disease phenotype. The gene corrected iPS cell derived haematopoietic progenitors showed stable engraftment and correction of disease phenotype.
• In another study from Jaenish group Werniig et al. derived dopaminergic neurons from iPS cells that, when implanted in the brain, became functionally integrated and improved rat model of Parkinson’s disease [89]. Thus these studies provide proof of principle for using reprogramming with gene repair and self-replacement therapy for treating disease. This is not compounded by the use of immunosuppressive drugs to prevent tissue rejection, while harnessing targeted gene repair strategies e.g. homologous recombination and zinc finger nucleases, to repair genetic defects. Unlimited population of stem cells can be differentiated into desired types by these strategies, for studying disease mechanisms, screening and developing drugs or for developing a suitable cell replacement therapy. It is still unclear if this would completely evade immune response with immune rejection of teratomas formed from iPS cells even in syngeneic mice [90].
Byrne et al. reviewed the advances in neural stem cells (NSC) as well as human induced pluripotent stem cells (iPSC’s) could provide a cure for neurodegenerative diseases like Alzheimer’s disease.
Recent discovery by Blurton Jones that NSC’s can effectively deliver disease modifying therapeutic proteins throughout the brain in murine brain [91] of best of model of AD studied. Other recent advances include that neprilysin, is a rate limiting enzyme in the degradation of amyloid β (Aβ) which was tried by a viral approach. In humans NSC’s offer a potential advantage to overcome this bottleneck of viral delivery by viral vectors. However the problem is that commonly used human whole fetal grafts [92] or NSC’s derived from fetal tissue would require immunosuppression, not being matched with the patient immunologically. Immunosuppressive drugs are expensive, inconvenient and not ideal. The alternate approach used is autologous NSC’s from human iPSC’s which themselves have been derived from suitable patient cells like skin cells (Figure 2). These cells could be modified in a manner similar to the NSC modification process implemented by Burton Jones. Problem of using these viral vectors is that they may induce insertional mutagenesis in a subset of cells [93]. One solution for this is use of flexible inexpensive genome editing techniques like CRISPR/Cas9 systems for targeting hiPSC. Also recent reports indicated that implanted iPSC derived NSC have demonstrated the ability to survive, migrate and differentiate and restore lost neurological function [94,95]. Thus, they summed up that with the pace of stem cell based regenerative therapies soon all these hurdles will be overcome and we will have answers for human neurodegenerative diseases [96].
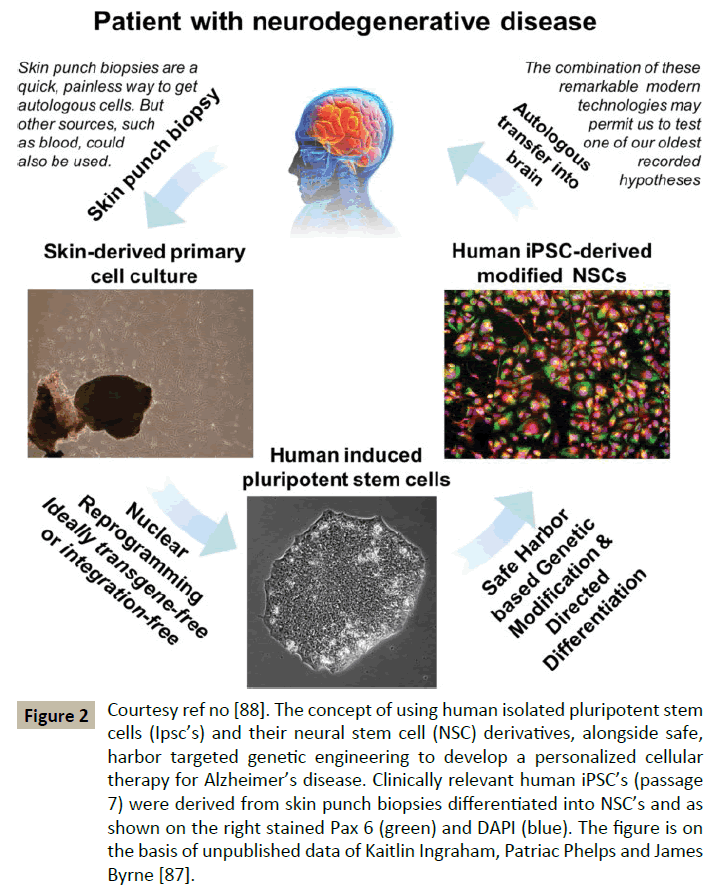
Figure 2: Courtesy ref no [88]. The concept of using human isolated pluripotent stem cells (Ipsc’s) and their neural stem cell (NSC) derivatives, alongside safe, harbor targeted genetic engineering to develop a personalized cellular therapy for Alzheimer’s disease. Clinically relevant human iPSC’s (passage 7) were derived from skin punch biopsies differentiated into NSC’s and as shown on the right stained Pax 6 (green) and DAPI (blue). The figure is on the basis of unpublished data of Kaitlin Ingraham, Patriac Phelps and James Byrne [87].
Role of Genome Editing Technologies Including CASPR/Cas System
Clustered regulatory interspersed short palindromic repeats (CRISPR/Cas) system is one of the genome editing technologies besides zinc finger nuclease (ZFN), Transcription activator like effector nucleases (TALEN). ZFN has gone upto clinical research stage in AIDS therapy based on administration of human chemokine like receptor5 (CCR5) modified T cells [97]. Gene correction by ZFN’s has also been reported in iPSC derived somatic cell biopsies in patients with sickle cell disease, α1 antitrypsin deficiency and Parkinson’s disease [98-101]. CRISPR/ Cas9 has been used to correct mutation in intestinal stem cells derived from patients with cystic fibrosis [102]. Two reports further cited that microinjection of Cas9 and TALEN into 1 cell stage embryos lead to efficient generation of targeted gene modified from nonhuman primates (NHP). In genome editing of mammals targeted gene modification is frequently carried out by microinjecting of gene editing system which consists of the nuclease mRNA, single guide RNAs (sgRNA’s for Cas9 and a homology containing donor DNA template (if necessary) into animal embryos made by IVF or ICSI [103-116]. Mammalian ESC’s, including human ESC’ shave been more efficiently modified by genome editing [107,108,117-120]. Genome editing 0technology is more likely to come into use in medicine for preventing a genetic disease if corrective genome editing is integrated into ART including IVF and ICSI. One important difference is that genome editing does not require cell donation e.g. oocyte donation as in ooplasmic transfer and mitochondrial replacement.
Mechanism-Genome editing technologies is more efficient genetic engineering which can directly modify a gene within a genome in various organisms. This is obtained by a microorganism–originated, engineered nuclease, which causes double stranded breaks (DSB’s) at a targeted sequence and induces DNA repair through non-homologous end joining (NHEJ) or homology directed repair (HDR). The NHEJ is a DSB repair pathway, which ligates or joins 2 broken ends together without a homologous template for repair and thus leading to the introduction of small insertions or deletions (indels) at the site of the DSB. The HDR is a DNA template dependent pathway for DSB repair, using a technology containing donor template along with a site specific genome editing nuclease, enabling the insertion of single or multiple transgenes (gene addition) in addition to single nucleotide substitutions in which an amino acid substitution of a protein occurs (gene modification), or a mutation is completely repaired in the resultant organism genome (gene corrections). Indications of integrating corrective genome editing into ART those with congenital anomalies, which are caused by chromosomal, monogeneic, multifactorial, or environmental/teratogenic factors [121]. Hence genome editing would maybe beneficial for monogeneic disease since a genome editing can efficiently repair such a small mutation in the human germline. But, use of genome editing for preventing transmission of a monogeneic disease should be limited to cases where the medical benefits exceed the potential health risks associated with the genetic intervention, implying definitive inheritance by the offspring e.g. autosomal recessive disease in which both parents are homozygous e.g. cystic fibrosis [122], phenylketonuria [123] or an autosomal dominant disease, where at least one parent is homozygous (e.g. Huntington’s disease, familial adenomatous polyposis [124] is likely to be considered.
If one attempts to repair a mutation directly in oocytes or embryos by means of an older homologous recombination technique, this attempt is likely to fail because of its low efficiency. Hence genome editing mediated gene corrections in ESC’s which are derived from a parents embryo made by IVF/ICSI could represent an alternative approach. Advantage of self-renewal of ESC’s, in vitro expansion and cryopreservation of ESC’s helps to repeatedly correct a mutation in a specific gene by genome editing. The efficiency of indel and gene addition is 14% to 91% by Cas9 and 0-83. 49% by ZFN or TALEN’s respectively.
Role in Gamete Generation
Hendricks et al. reviewed the advances in generation of oocytes and sperms by manipulation of the progenitor cells and somatic cells in lieu of shortage of donor gametes and ethicality involved [125]. Valli et al. further reviewed various modalities of obtaining spermatogonial stem cells, SSC for transplantation, testicular homografting organ culture in prepubertal boys where gametes are not mature enough to be preserved by cryopreservation before gonadotoxic chemotherapy in malignancies and various procedures of generation of germ cells from pluripotent stem cells iPSC’s though the function of resultant germ cells has not been possible to test in human system although with ongoing advances in technology days are not far when such men will have their own gametes with the persistent shortage of gametes, both oocytes and sperms [126].
References
- Evans MJ, Kaufman MH (1981) Establishment in culture of pluripotential cells from mouse embryos. Nature 292: 154-156.
- Schwartz SD, Hubschman JP, Heilwell G, Franco-Cardenas V, Pan CK, et al. (2012) embryonic stem cell trials for macular degeneration: a preliminary report. Lancet 379: 713-720.
- https://clinicaltrials.gov/ct2/show/NCT01469832
- Abbondanzo SJ, Gadi I, Stewart CL (1993) Derivation of embryonic stem cell lines. Methods Enzymol 225: 803-823.
- Hogan P, Beddington R, Constantini F, Lacy F (1994) Isolation, culture and manipulation of embryonic stem cells (2ndedn) In Manipulating the mouse embryo. A Laboratory Manual, Cold Spring Harbor Labortory, Press, New York Pp: 254-290.
- Suemori H, Nakatsuji N (1987) Establishment of the embryo derived stem ES cell lines from mouse blastocysts: effects of feeder layer. Dev Growth Differ 29: 133-139.
- Stewart CL, Kaspar P, Brunet LJ, Bhatt H, Gadi I, et al. (1992) Blastocyst implantation depends on maternal expression of leukaemia inhibitory factor. Nature 359: 76-79.
- Pease S, Braghetta P, Gearing D, Grail D, Williams RL (1990) Isolation of embryonic stem ES cells in media supplemented with recombinant leukemia inhibitory factor. Dev Biol 141: 344-352.
- Coroner JC, Ip NY, Poueymirou WT, Bates B, Goldfarh B, et al. (1993) Ciliaryneutrophic factor maintains the pluripotentiality of embryonic sytem cell lines from human blastocyst. Development 119: 559-565.
- Rose TM, Weiford DM, Gunderson NL, Bruce AG (1994) Oncostatin M (OSM) inhibits the differentiation of pluripotent embryonic stem cells in vitro. Cytokine 6: 48-54.
- Nichols J, Chambers I, Smith A (1994) Derivation of germline competent embryonic stem cells with a combination of interleukin-6 and soluble interleukin-6 receptor. Exp Cell Res 215: 237-239.
- Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, et al. (1998) Embryonic stem cell lines derived from human blastocysts. Science 282: 1145-1147.
- Martin MJ, Muotri A, Gage F, Varki A (2005) Human embryonic stem cells express an immunogenic nonhuman sialic acid. Nat Med 11: 228-232.
- Xu C, Inokuma MS, Denham J, Golds K, Kundu P, et al. (2001) Feeder-free growth of undifferentiated human embryonic stem cells. Nat Biotechnol 19: 971-974.
- Draper JS, Moore HD, Ruban LN, Gokhale PJ, Andrews PW (2004) Culture and characterization of human embryonic stem cells. Stem Cells Dev 13: 325-336.
- Cowan CA, KlimanskayaI, McMohan J, Alienza J, Wilmyer J, et al. (2004) Derivation of embryonic stem cell lines from human blastocysts. N Engl J Med 350: 1353-1356.
- Hwang WS, Ryu YJ, Park JH, Park ES, Lee EG, et al. (2004) Evidence of a pluripotent human embryonic stem cell line derived from a cloned blastocyst. Science 303(5664): 1669-1674.
- Cibelli JB, Lanza RP, West MD, Ezzell C (2001) Somatic cell nuclear transfer in human: Pronuclear and early embryonic development. J Regen Med 26: 25-31.
- Hwang WS, Roh SI, Lee BC, Kang SK, Kwon DK, et al. (2005) Patient-specific embryonic stem cells derived from human SCNT blastocysts. Science 308(5729): 1777-1783.
- Humpherys D, Eggan K, Akutsu H, Friedman A, Hochedlinger K, et al. (2002) Abnormal gene expression in cloned mice derived from embryonic stem cell and cumulus cell nuclei. Proc Natl Acad Sci USA 99: 12889-12894.
- St John JC, Schatten G (2004) Paternal mitochondrial DNA transmission during nonhuman primate nuclear transfer. Genetics 167: 897-905.
- Lanza RP, Chung HY, Yoo JJ, Wettstein PJ, Blackwell C, et al. (2002) Generation of histocompatible tissues using nuclear transplantation. Nat Biotechnol 20: 689-696.
- Rideout WM, Hochedlinger K, Kyba M, Daley GQ, Jaenisch R (2002) Correction of a genetic defect by nuclear transplantation and combined cell and gene therapy. Cell 109: 17-27.
- Barberi T, Klivenyi P, Calingasan NY, Lee H, Kawamata H, et al. (2003) Neural subtype specification of fertilization and nuclear transfer embryonic stem cells and application in parkinsonian mice. Nat Biotechnol 21: 1200-1207.
- Crews D, Grassman M, Lindzey J (1986) Behavioral facilitation of reproduction in sexual and unisexual whiptail lizards. Proc Natl Acad Sci USA 83: 9547-9550.
- Chapman DD, Shivji MS, Louis E, Sommer J, Fletcher H, et al. (2007) Virgin birth in a hammerhead shark. Biol Lett 3: 425-427.
- Kono T, Obata Y, Wu Q, Niwa K, Ono Y, et al. (2004) Birth of parthenogenetic mice that can develop to adulthood. Nature 428: 860-864.
- Kline D, Kline JT (1992) Repetitive calcium transients and the role of calcium in exocytosis and cell cycle activation in the mouse egg. Dev Biol 149: 80-89.
- Kashir J, Nomikos M, Swann K, Lai FA (2015) PLCζ or PAWP: revisiting the putative mammalian sperm factor that triggers egg activation and embryogenesis. Mol Hum Reprod 21: 383-388.
- Bos-Mikich A, Whittingham DG, Jones KT (1997) Meiotic and mitotic Ca2+ oscillations affect cell composition in resulting blastocysts. Dev Biol 182: 172-179.
- BosMikich A, Swann K, Whittingham DG (1995) Calcium oscillations and protein synthesis inhibition synergistically activate m ouseoocytes. Molecular Reproduction and Development 41: 84-90.
- Ruggeri RR, Watanabe Y, Meirelles F, Bressan FF, Frantz N, et al. (2012) The use of parthenotegenetic and IVF bovine blastocysts as a model for the creation of human embryonic stem cells under defined conditions. J Assist Reprod Genet 29: 1039-1043.
- Ruggerri RR, Bressan FF, Sujeira NM, Meirelles F, Frantz N, et al. (2014) Derivation and culture of putative parthenogenetic embryonic stem cells in new gelatin substrates modified with galactomannan. Macromolecular Research 22: 1053-1058.
- Mitalipov SM, Nusser KD, Wolf DP (2001) Parthenogenetic activation of rhesus monkey oocytes and reconstructed embryos. Biol Reprod 65: 253-259.
- Susko-Parrish JL, Leibfried-Rutledge ML, Northey DL, Schutzkus V, First NL (1994) Inhibition of protein kinases after an induced calcium transient causes transition of bovine oocytes to embryonic cycles without meiotic completion. Dev Biol 166: 729-739.
- Brevini TA, Gandolfi F (2008) Parthenotes as a source of embryonic stem cells. Cell Prolif 41(1): 20-30.
- Mai Q, Yu Y, Li T, Wang L, Chen MJ, et al. (2007) Derivation of human embryonic stem cell lines from parthenogenetic blastocysts. Cell Res 17: 1008-1019.
- Tachibana M, Amato P, Sparman M, Woodward J, Sanchis DM, et al. (2013) Towards germline gene therapy of inherited mitochondrial diseases. Nature 493: 627-631.
- Tachibana M, Sparman M, Sritanaudomchai H, Ma H, Clepper L, et al. (2009). Mitochondrial gene replacement in primate offspring and embryonic stem cell. Nature 461: 367-372.
- Revazova ES, Turovets NA, Kochetkova OD, Kindarova LB, Kuzmichev LN, et al. (2007) Patient-specific stem cell lines derived from human parthenogenetic blastocysts. Cloning Stem Cells 9: 432-449.
- Park KS, SongHB, Chun SS (2000) Late fertilization of unfertilized human oocytes in invitro fertilization and intracytoplasmic sperm injection cycles: conventional insemination vs ICSI. J Assist Reprod Genet 17: 419-424.
- Kuczyski W, Dhont M, Grygoruk C, Pietrewicz P, Redzko S, et al. (2002) Rescue ICSI of unfertilized oocytes after IVF. Hum Reprod 17: 2423-2427.
- Lombardi L, Tiveron M, Inza A, Valcrcel A, Young E, Bisioloi C (2003) Live birth and normal 1 year follow up of a baby born after transfer of cryopreserved embryos from rescue intracytoplasmic injection of 1 day old oocyte. Fertil Steril 80: 646-648.
- Amarin ZO, Obeidat BR, Rouzi AA, Jallad MF, Khader YS (2005) Intracytoplasmic sperm injection after total conventional in-vitro fertilization failure. Saudi Med J 26: 411-415.
- Sneddon SF, DeSousa PA, Arnesen RE, Lieberman BA, Kimber SJ, et al. (2011) Gene expression analysis of a new source of human oocytes and embryos for research and human embryonic stem cell derivation. Fertil Steril 95: 1410-1415.
- Kimber SJ, Sneddon SF, Bloor DJ, El-Bareg AM, Hawkhead JA, et al. (2008) Expression of genes involved in early cell fate decisions in human embryos and their regulation by growth factors. Reproduction 135: 635-647.
- Kyono K, Kumagai S, Nishinaka C, Nakajo Y, Uto H, et al. (2008) Birth and follow-up of babies born following ICSI using SrCl2 oocyte activation. Reprod Biomed Online 17: 53-58.
- Kyono K, Nakajo Y, Nishinaka C, Hattori H, Kyoya T, et al. (2009) A birth from the transfer of a single vitrified warmed blastocyst using intracytoplasmic sperm injection with calcium activation in a globozoospermicpatient. Fertil Steril 91: e7-e11.
- Murase Y, Araki S, Mizuno S, Kawaguchi C, Naito M, et al. (2004) Pregnancy following chemical activation of oocytes in a couple with repeat filure of fertilization using ICSI:A Case Report. Hum Reprod 19: 1604-1607.
- Pinto J, Check JH (2008) Correction of failed fertilization despite intractyoplsmic sperm injection with oligoasthenozoospermia but with acrosome present by oocyte activation with calcium ionophore-case report. Clin Exp Obstet Gynecol 35: 252-254.
- Borges E Jr, Braga PD, de Sousa Bonetti TC (2009) Artificial oocyt activation with calcium ionophorein ICSI cycles with spermatozoa from different sources. Reproductive Biomedicine Online 18: 45-52.
- Check JH, Summers-Chase D, Cohen R, Brasik D (2010) Artificial oocyte activation with calcium ionophore allowed fertilization and pregnancy in a couple with long term unexplained infertility where the female partner had diminished egg reserve and failure to fertilize oocytes despite intracytoplasmic injection. Clin Exp Obstet Gynecol 37: 263-265.
- Kono T (2006) Genomic imprinting is a barrier to parthenogenesis in mammals. Cytogenet Genome Res 113: 31-35.
- Kaneko-Ishino T, Kohda T, Ishino F (2003) The regulation and biological significance of genomic imprinting in mammals. J Biochem 133: 699-711.
- Sritanaudomchai H, Ma H, Clepper L, Gokhale S, Bogan R, et al. (2010) Discovery of a novel imprinted gene by transcriptional analysis of parthenogenetic embryonic stem cells. Hum Reprod 25: 1927-1941.
- Surani MA, Barton SC, Norris ML (1984) Development of reconstituted mouse eggs suggests imprinting of the genome during gametogenesis. Nature 308: 548-550.
- Barlow DP, Stoger R, Herrmann BG, Saito K, Schweifer N (1991) The mouse insulin-like growth factor type-2 receptor is imprinted and closely linked to the Tme locus. Nature 349: 84-87.
- DeChiara TM, Robertson EJ, Efstratiadis A (1991) Parental imprinting to the mouse insulin like growth factor II gene. Cell 64: 849-859.
- Ferguson-Smith AC, Cattanach BM, Barton SC, Beechey CV, Surani MA (1991) Embryological and molecular investigations of parental imprinting on mouse chromosome 7. Nature 351: 667-670.
- Bartolomei MS, Zemel S, Tilghman SM (1991) Parental imprinting of the mouse H19 gene. Nature 351: 153-155.
- Ogawa H, Shindo N, Kumagai T, Usami Y, Shikanai M, et al. (2009) Developmental ability of trophoblast stem cells in uniparental mouse embryos. Placenta 30: 448-456.
- Obata Y, Kaneko-IshinoT, Koide T, Takai Y, Ueda T, et al. (1998) Disruption of primary imprinting during oocyte growth leads to the modified expression of imprinted genes during embryogenesis. Development 125: 1553-1560.
- Constância M, Hemberger M, Hughes J, Dean W, Ferguson-Smith A, et al. (2002) Placental-specific IGF-II is a major modulator of placental and fetal growth. Nature 417: 945-948.
- Kawahara M, Wu Q, Takahashi N, Morita S, Yamada K, et al. (2007) High-frequency generation of viable mice from engineered bi-maternal embryos. Nat Biotechnol 25: 1045-1050.
- Kawahara M, Kono T (2010) Longevity in mice without a father. Hum Reprod 25: 457-461.
- Jiang YH, Bressler J, Beaudet AL (2004) Epigenetics and human disease. Annu Rev Genomics Hum Genet 5: 479-510.
- Uh KJ, Park CH, Choi KH, Park JK, Jeong YW, et al. (2014) Analysis of imprinted IGF2/H19 gene methylation and expression in normal fertilized and parthenogenetic embryonic stem cells of pigs. Anim Reprod Sci 147: 47-55.
- Didié M, Christalla P, Rubart M, Muppala V, Döker S, et al. (2013) Parthenogenetic stem cells for tissue-engineered heart repair. J Clin Invest 123: 1285-1298.
- Streckfuss-Bömeke K, Wolf F, Azizian A, Stauske M, Tiburcy M, et al. (2012) Comparitive study of human induced pluripotent stem cells derived from bone marrow cells, hair keratinocytes and skin fibroblasts. Eur Heart J 34: 2618-2629.
- Soong PL, Tiburcy M, Zimmermann WH (2012) Cardiac differentiation of human embryonic stem cells and their assembly into engineered heart muscle. Curr Protoc Cell Biol Chapter 23: Unit23.
- Byrne JA, Pedersen DA, Clepper LL, Nelson M, Sanger WG, et al. (2007) Producing primate embryonic stem cells by somatic cell nuclear transfer. Nature 450: 497-502.
- Cibelli JB, Grant KA, Chapman KB, Cunniff K, Worst T, et al. (2002) Parthenogenetic stem cells in nonhuman primates. Science 295: 819.
- Vrana KE, Hipp JD, Goss AM, McCool BA, Riddle DR, et al. (2003) Nonhuman primate parthenogenetic stem cells. Proc Natl Acad Sci U S A 100 Suppl 1: 11911-11916.
- Barton SC, Surani MA, Norris ML (1984) Role of paternal and maternal genomes in mouse development. Nature 311: 374-376.
- Nakagawa H, Chadwick RB, Peltomaki P, Plass C, Nakamura Y, et al. (2001) Loss of imprinting of the insulin-like growth factor II gene occurs by biallelic methylation in a core region of H19-associated CTCF-binding sites in colorectal cancer. Proc Natl Acad Sci U S A 98: 591-596.
- Dighe V, Clepper L, Pedersen D, Byrne J, Ferguson B, et al. (2008) Heterozygous embryonic stem cell lines derived from nonhuman primate parthenotes. Stem Cells 26: 756-766.
- Kim K, Lerou P, Yabuuchi A, Lengerke C, Ng K, et al. (2007) Histocompatible embryonic stem cells by parthenogenesis. Science 315: 482-486.
- Paull D, Emmanuele V, Weiss KA, Treff N, Stewart L, et al. (2013) Nuclear genome transfer in human oocytes eliminates mitochondrial DNA variants. Nature 493: 632-637.
- Espejel S, Eckardt S, Harbell J, Roll GR, McLaughlin KJ, et al. (2014) Brief report: Parthenogenetic embryonic stem cells are an effective cell source for therapeutic liver repopulation. Stem Cells 32: 1983-1988.
- Eckardt S, Leu NA, Bradley HL, Kato H, Bunting KD, et al. (2007) Hematopoietic reconstitution with androgenetic and gynogenetic stem cells. Genes Dev 21: 409-419.
- Sánchez-Pernaute R, Studer L, Ferrari D, Perrier A, Lee H, et al. (2005) Long-term survival of dopamine neurons derived from parthenogenetic primate embryonic stem cells (cyno-1) after transplantation. Stem Cells 23: 914-922.
- Lee G, Papapetrou EP, Kim H, Chambers SM, Tomishima MJ, et al. (2009) Modelling pathogenesis and treatment of familial dysautonomia using patient-specific iPSCs. Nature 461: 402-406.
- Moretti A, Bellin M, Welling A, Jung CB, Lam JT, et al. (2010) Patient-specific induced pluripotent stem-cell models for long-QT syndrome. N Engl J Med 363: 1397-1409.
- Itzhaki I, Maizels L, Huber I, Zwi-Dantsis L, Caspi O, et al. (2011) Modelling the long QT syndrome with induced pluripotent stem cells. Nature 471: 225-229.
- Agarwal S, Loh YH, McLoughlin EM, Huang J, Park IH, et al. (2010) Telomere elongation in induced pluripotent stem cells from dyskeratosis congenita patients. Nature 464: 292-296.
- Batista LF, Pech MF, Zhong FL, Nguyen HN, Xie KT, et al. (2011) Telomere shortening and loss of self-renewal in dyskeratosis congenita induced pluripotent stem cells. Nature 474: 399-402.
- Aggarwal S, Daley GQ (2011) Telomere dynamics in dyskeratosis congenital: the long and the short of iPS. Cell Res.
- Hanna J, Wernig M, Markoulaki S, Sun CW, Meissner A, et al. (2007) Treatment of sickle cell anemia mouse model with iPS cells generated from autologous skin. Science 318: 1920-1923.
- Wernig M, Zhao JP, Pruszak J, Hedlund E, Fu D, et al. (2008) Neurons derived from reprogrammed fibroblasts functionally integrate into the fetal brain and improve symptoms of rats with Parkinson's disease. Proc Natl Acad Sci U S A 105: 5856-5861.
- Zhao T, Zhang ZN, Rong Z, Xu Y (2011) Immunogenicity of induced pluripotent stem cells. Nature 474: 212-215.
- Blurton Jones M, Spencer B, Mitchell S, Castello NA, Agazaryan AA, et al. (2014) Neural stem cells genetically modified to express neprilysin reduce pathology in Alzheimers transgenic models. Stem Cell Res 5: 46.
- Freed CR, Breeze RE, Rosenberg NE, Schneck SA, Kriek E, et al. (1992) Survival of implanted fetal dopamine cells and neurologic improvement 12 to 46months after transplantation for parkinsons disease. N Engl J Med 327: 1549-1555.
- Byrne JA (2013) Resolving clinical hurdles for autologous pluripotent stem cell based therapies. OA Stem Cells 1: 3.
- Awe JP, Lee PC, Ramathal C, Vega-Crespo A, Durruthy-Durruthy J, et al. (2013) Generation and characterization of transgene-free human induced pluripotent stem cells and conversion to putative clinical-grade status. Stem Cell Res Ther 4: 87.
- Yuari T, Liao W, Feng NH, Looyl, Niu X, et al. (2013) Human induced pluripotent stem cell derived neural stem cell survival migrate ,differentiate ,and improve technological functions in a rat model of middle cerebral artery occlusion. Stem Cell Res 4: 23.
- Byrne JA (2014) Developing neural stem cell-based treatments for neurodegenerative diseases. Stem Cell Res Ther 5: 72.
- Tebas P, Stein D, Tang WW, Frank I, Wang SQ, et al. (2014) Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. N Engl J Med 370: 901-910.
- Sebastiano V, Maeder ML, Angstman JF, Haddad B, Khayter C, et al. (2011) In situ genetic correction of the sickle cell anemia mutation in human induced pluripotent stem cells using engineered zinc finger nucleases. Stem Cells 29: 1717-1726.
- Yusa K, Rashid ST, Strick-Marchand H, Varela I, Liu PQ, et al. (2011) Targeted gene correction of α1-antitrypsin deficiency in induced pluripotent stem cells. Nature 478: 391-394.
- Soldner F, Laganire J, Cheng AW, Hockemeyer D, Gao Q, et al. (2011) Generation of isogenic pluripotent stem cells differing exclusively at two early onset Parkinson point mutations. Cell 146: 318-331.
- Schwank G, Koo BK, Sasselli V, Dekkers JF, Heo I, et al. (2013) Functional repair of CFTR by CRISPR/Cas9 in intestinal stem cell organoids of cystic fibrosis patients. Cell Stem Cell 13: 653-658.
- Bacnan SR,Williams SI, Pinto M, Peralta S, Moraes CT (2013) Specific elimination of mutant mitochondrial genomes in patients derived by mitoTALEN’s. Nat Med 19: 1111-1113.
- Liu H, Chen Y, Niu Y, Zhang K, Kang Y, et al. (2014) TALEN-mediated gene mutagenesis in rhesus and cynomolgus monkeys. Cell Stem Cell 14: 323-328.
- Niu Y, Shen B, Cui Y, Chen Y, Wang J, et al. (2014) Generation of gene-modified cynomolgus monkey via Cas9/RNA-mediated gene targeting in one-cell embryos. Cell 156: 836-843.
- Wu Y, Liang D, Wang Y, Bai M, Tang W, et al. (2013) Correction of a genetic disease in mouse via use of CRISPR-Cas9. Cell Stem Cell 13: 659-662.
- Tesson L, Usal C, MÃnoret S, Leung E, Niles BJ, et al. (2011) Knockout rats generated by embryo microinjection of TALENs. Nat Biotechnol 29: 695-696.
- Yang H, Wang H, Shivalila CS, Cheng AW, Shi L, et al. (2013) One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell 154: 1370-1379.
- Li W, Teng F, Li T, Zhou Q (2013) Simultaneous generation and germline transmission of multiple gene mutations in rat using CRISPR-Cas systems. Nat Biotechnol 31: 684-686.
- Ran FA, Hsu PD, Lin CY, Gootenberg JS, Konermann S, et al. (2013) Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 154: 1380-1389.
- Wang B, Yang H, Shivalla CS, Dawlaty MM, Cheng AW, et al. (2013) One step generation of mice carrying mutations in multiple genes by CRISPR/Cas mediated genome engineering. Cell 153: 910-918.
- Li F, Cowley DO, Banner D, Holle E, Zhang L, et al. (2014) Efficient genetic manipulation of the NOD-Rag1-/-IL2RgammaC-null mouse by combining in vitro fertilization and CRISPR/Cas9 technology. Sci Rep 4: 5290.
- Yasue A, Mitsui SN, Watanabe T, Sakuma T, Oyadomari S, et al. (2014) Highly efficient targeted mutagenesis in one-cell mouse embryos mediated by the TALEN and CRISPR/Cas systems. Sci Rep 4: 5705.
- Carlson DF, Tan W, Lillico SG, Stverakova D, Proudfoot C, et al. (2012) Efficient TALEN-mediated gene knockout in livestock. Proc Natl Acad Sci U S A 109: 17382-17387.
- Liu Z, Zhou X, Zhu Y, Chen ZF, Yu B, et al. (2014) Generation of a monkey with MECP2 mutations by TALEN-based gene targeting. Neurosci Bull 30: 381-386.
- Mashimo T, Kaneko T, Sakuma T, Kobayashi J, Kunihiro Y, et al. (2013) Efficient gene targeting by TAL effector nucleases coinjected with exonucleases in zygotes. Sci Rep 3: 1253.
- Lombardo A, Genovese P, Beausejour CM, Colleoni S, Lee YL, et al. (2007) Gene editing in human stem cells using zinc finger nucleases and integrase-defective lentiviral vector delivery. Nat Biotechnol 25: 1298-1306.
- Hockemeyer D, Soldner F, Beard C, Gao Q, Mitalipova M, et al. (2009) Efficient targeting of expressed and silent genes in human ESCs and iPSCs using zinc-finger nucleases. Nat Biotechnol 27: 851-857.
- Hockemeyer D, Wang H, Kiani S, Lai CS, Gao Q, et al. (2011) Genetic engineering of human pluripotent cells using TALE nucleases. Nat Biotechnol 29: 731-734.
- Zou J, Maeder ML, Mali P, Pruett-Miller SM, Thibodeau-Beganny S, et al. (2009) Gene targeting of a disease-related gene in human induced pluripotent stem and embryonic stem cells. Cell Stem Cell 5: 97-110.
- Li W, Li X, Li T, Jiang MG, Wan H, et al. (2014) Genetic modification and screening in rat using haploid embryonic stem cells. Cell Stem Cell 14: 404-414.
- WHO (2014) Congenital Anomales. Fact Sheet number 370.
- Neocleous V, Yiallouros PK, Tanteles GA, Costi C, Moutafi M, et al. (2014) Apparent Homozygosity of p.Phe508del in CFTR due to a Large Gene Deletion of Exons 4-11. Case Rep Genet 2014: 613863.
- Gro Selj U, Tansel MZ, Kovak J, Hovil D, Podkrajsek KT, et al. (2012) Five novel mutations and two large deletions in in a population analysis of the phenylalanine hydroxylase gene. Mol Genet Metab 106: 142-148.
- Cruz-Correa M, Diaz-Algorri Y, Mendez V, Vasquez PJ, Lozada ME, et al. (2013) Clinical characterization and mutation spectrum in Hispanic families with adenomatous polyposis syndrome. Familial Cancer 12: 555-562.
- Hendriks S, Dancet EA, van Pelt AM, Hamer G, Repping S (2015) Artificial gametes: a systematic review of biological progress towards clinical application. Hum Reprod Update 21: 285-296.
- Valli H, Phillips BT, Shetty G, Byrne JA, Clark AT, et al. (2014) Germline stem cells: toward the regeneration of spermatogenesis. Fertil Steril 101: 3-13.